Bioinformatics Pipeline
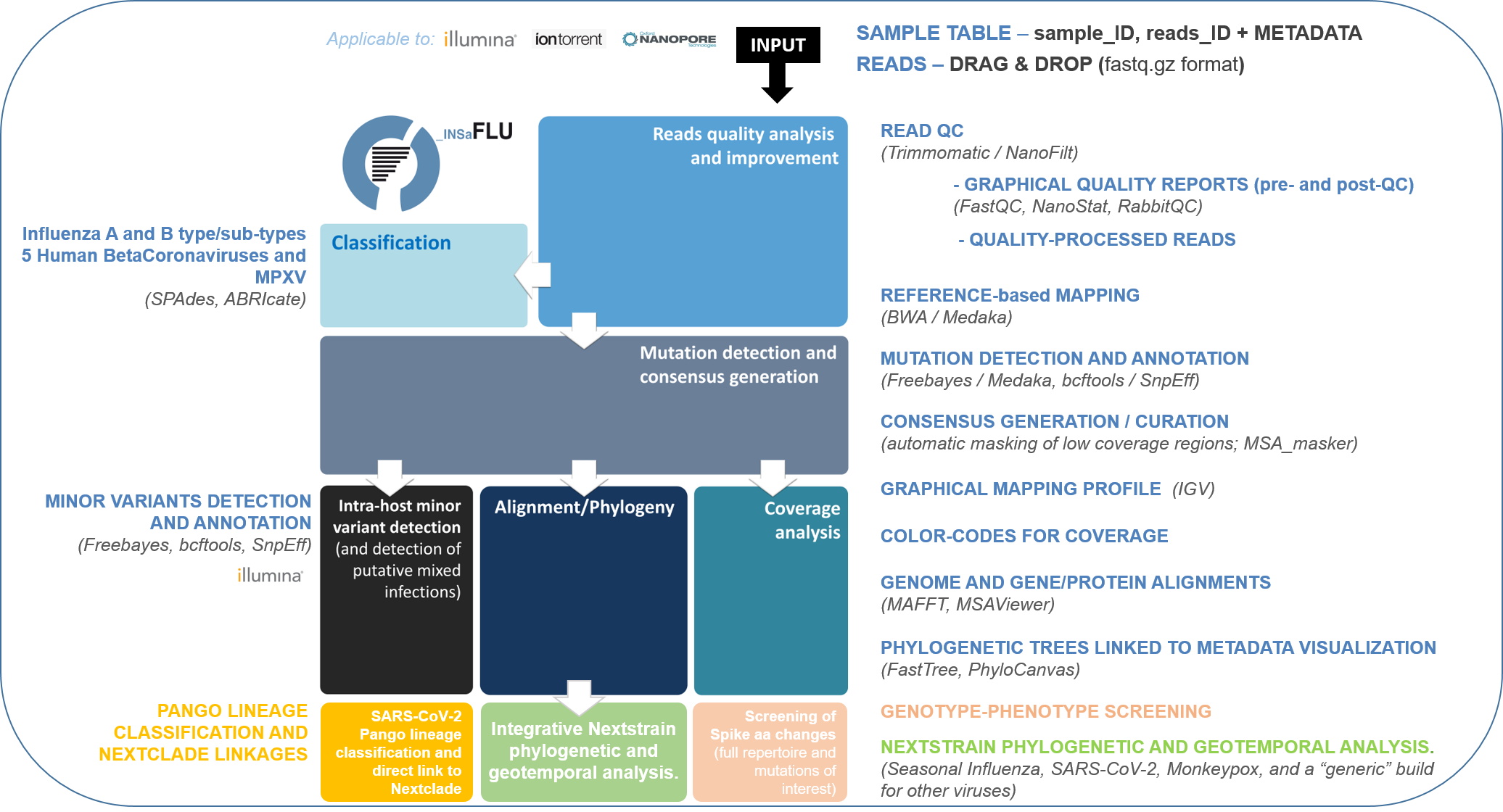
INSaFLU is an open web-based bioinformatics platform for metagenomic virus detection and routine genomic surveillance.
As such, it integrates two main pipelines / components:
a Reference-based genomic surveillance pipeline (from NGS reads to quality control, mutations detection, consensus generation, virus classification, alignments, “genotype-phenotype” screening, phylogenetics, integrative phylogeographical and temporal analysis etc)
a Virus detection pipeline (from NGS reads to quality control and metagenomics virus identification)
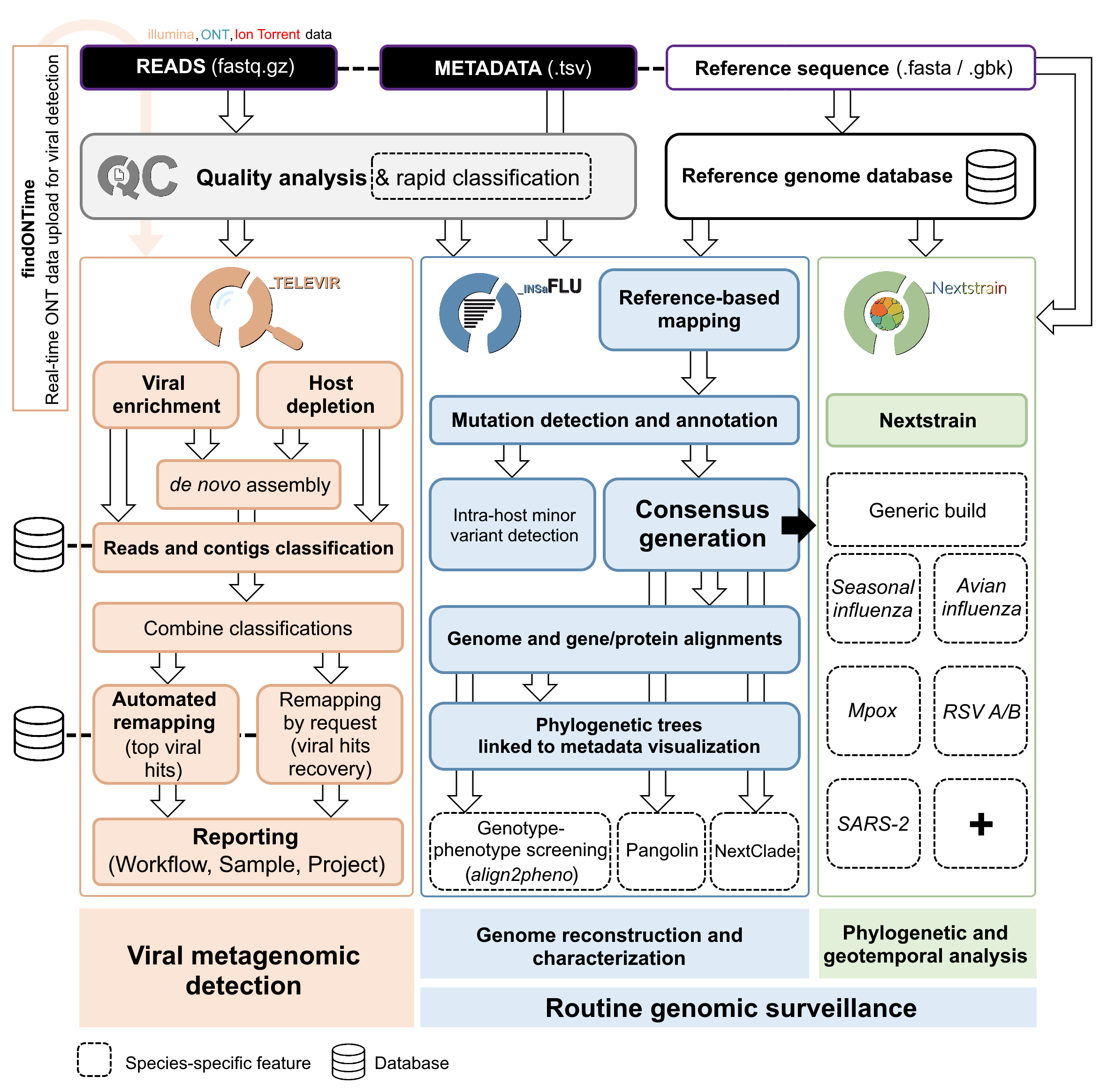
INSaFLU relies on a multi-software bioinformatics pipelines that will be under continuous development and improvement not only to enrich it with new features, but also to continuously shape the protocol to the best methodological advances in the field.
The current software and default settings, which were chosen upon intensive testing, are described below, together with the list of Steps and Settings that can be turned ON/OFF or configured by the user, respectively. For additional details about the bioinformatics pipeline, please visit the INSaFLU github account: https://github.com/INSaFLU/INSaFLU (more information for each software can also be found in the official repositories; links are also provided below).
Reference-based genomic surveillance
The “surveillance-oriented” bioinformatics component of INSaFLU allows launching:
Projects - From reads to reference-based generation of consensus sequences and mutations annotation, followed by gene- and genome-based alignments, amino acid alignments, Pango classification, NextClade linkage, etc.
and
Datasets - From consensus sequences to advanced Nextstrain phylogenetic and genomic analysis, coupled with geographic and temporal data visualization and exploration of sequence metadata.
The bioinformatics pipeline currently consists of multiple steps (see WorkFlow) yielding multiple graphical, and sequence outputs (see Output visualization and download menu for details)
Simplified illustration of the main steps of the modular INSaFLU-TELEVIR bioinformatics pipeline for reference-based genomics surveillance.
Find below the description of the main bioinformatics steps (Description and Current software )
Read quality analysis and improvement
Description
This step takes the input single- or paired-end reads (fastq.gz format) and produces quality processed reads, as well as quality control reports for each file, before and after quality improvement. This module is automatically run upon reads upload (i.e., no user intervention is needed).
Software version/settings
Note
## ILLUMINA / Ion Torrent data ##
FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) (version 0.11.5; date 15.01.2018)
input: single- or paired-end reads (fastq.gz format) (e.g., sample_L001_R1_001.fastq.gz and sample_L001_R2_001.fastq.gz for Illumina technology reads)
—nogroup option: all reports will show data for every base in the read.
Trimmomatic (http://www.usadellab.org/cms/index.php?page=trimmomatic) (version 0.27; date 15.01.2018)
input: single- or paired-end reads (fastq.gz format) (e.g., sample_L001_R1_001.fastq.gz and sample_L001_R2_001.fastq.gz for Illumina paired-end reads)
ILLUMINACLIP: To clip adapters from the input file using user-specified adapter sequences. ILLUMINACLIP:<ADAPTER_FILE>:3:30:10:6:true
HEADCROP: Cut the specified number of bases from the start of the read. Range: [0:100]. If value equal to 0 this parameter is excluded.
CROP: Cut the read to a specified length. Range: [0:400]. If value equal to 0 this parameter is excluded.
SLIDINGWINDOW: perform a sliding window trimming, cutting once the average quality within the window falls below a threshold (default: SLIDINGWINDOW:5:20, where 5 refers to window and 20 to the minimum average quality)
LEADING: cut bases off the start of a read, if below a threshold quality (default: LEADING:3). This will allow discarding bases with very quality or N bases (quality score of 2 or less).
TRAILING: cut bases off the end of a read, if below a threshold quality (default: TRAILING:3). This will allow discarding bases with very quality or N bases (quality score of 2 or less).
MINLEN: drop the read if it is below a specified length (MINLEN:35)
TOPHRED33: Convert quality scores to Phred-33
## Oxford Nanopore Technologies (ONT) data ##
NanoStat (https://github.com/wdecoster/nanostat) (version 1.4.0)
input: ONT reads (fastq.gz format)
NanoFilt (https://github.com/wdecoster/nanofilt) (version 2.6.0)
-q (QUALITY): Filter on a minimum average read quality score. Range: [5:30] (default: 10)
-l (LENGTH): Filter on a minimum read length. Range: [50:1000]. (default: 50)
–headcrop: Trim n nucleotides from start of read. Range: [1:1000]. If value equal to 0 this parameter is excluded. (default: 70)
–tailcrop: Trim n nucleotides from end of read. Range: [1:1000]. If value equal to 0 this parameter is excluded. (default: 70)
–maxlength: Set a maximum read length. Range: [100:50000]. If value equal to 0 this parameter is excluded. (default: 0)
RabbitQC (https://github.com/ZekunYin/RabbitQC) (version 0.0.1)**
input: ONT reads (fastq.gz format) pre- and post- quality improvement with NanoFilt
In the local installation, you can setup the env file to request that large files are downsized before analysis by randomly sampling reads. The tool fastq-sample from fastq-tools package https://github.com/dcjones/fastq-tools (developed by Daniel C. Jones dcjones@cs.washington.edu) is used for that purpose.
Note
## ILLUMINA data only ##
***Users can also use trimmomatic to perform trimming of primer sequences of several predefined Primer pool sets, including:
– SARS-CoV-2 Primal Scheme V3 (https://github.com/artic-network/artic-ncov2019/blob/master/primer_schemes/nCoV-2019/V3/nCoV-2019.tsv)
—SARS-CoV-2 Primal Scheme V4.1 (https://github.com/artic-network/artic-ncov2019/tree/master/primer_schemes/nCoV-2019/V4.1)
– Monkeypox Primal Scheme from Welkers, Jonges and van den Ouden (https://www.protocols.io/view/monkeypox-virus-whole-genome-sequencing-using-comb-n2bvj6155lk5/v1)
—Monkeypox Primal Scheme from Chen et al. (https://www.protocols.io/view/monkeypox-virus-multiplexed-pcr-amplicon-sequencin-5qpvob1nbl4o/v2)
Users can upload their own Primer sets (see https://insaflu.readthedocs.io/en/latest/uploading_data.html#uploading-primer-panels)
Important
INSaFLU allows users to configure key parameters for reads quality analysis in the tab “Settings”.
Please check your settings before uploading new samples to your account.
See details in https://insaflu.readthedocs.io/en/latest/data_analysis.html#user-defined-parameters
Variant detection and consensus generation
Description
This key module takes enables reference-based mapping, followed by SNP/indel calling and annotation and generation of consensus sequences (quality processed reads obtained through Trimmomatic analysis are used as input). Quality processed reads obtained through Trimmomatic (Illumina/IonTorrent data) NanoFilt (ONT data) are used as input. A reference sequence must be selected for each project (select one from INSaFLU default reference database or upload one of your choice). Uploaded “.fasta” files are annotated upon submission and automatically become available at the user-restricted reference database. For influenza, each project should ideally include viruses from the same type and sub-type/lineage (this typing data is automatically determined upon reads submission to INSaFLU).
Software version/settings
Note
##REFERENCE ANNOTATION##
Prokka (https://github.com/tseemann/prokka) (version 1.12; date 15.01.2018)
–kingdom: defines the Annotation mode (Viruses)
##ILLUMINA / Ion Torrent data##
Snippy (https://github.com/tseemann/snippy) (version 3.2-dev - sligthly modified (details in https://github.com/INSaFLU/INSaFLU); date 15.01.2018)
–R1 (and –R2): define the reads files used as input, i.e, quality processed reads (e.g., sample_1P.fastq.gz and sample_2P.fastq.gz) obtained after Trimmomatic analysis
—ref: define the reference sequence selected by the users (.fasta or gbk format)
–mapqual: minimum mapping quality to accept in variant calling(default: –mapqual 20)
—mincov: minimum coverage of variant site (default: –mincov 10)
–minfrac: minimum proportion for variant evidence (default: –minfrac 0.51)
—primer: defines primer sequences to be removed using iVar(version 1.4.2, available since 16/06/2023) (by default no primers are removed). The primer removal procedure was based on the iVar CookBook (https://github.com/andersen-lab/paper_2018_primalseq-ivar/blob/master/cookbook/CookBook.ipynb), but where no quality filtering is performed, and reads starting outside the primer are not excluded. Primer removal is obtained after the alignment step, but before variant calling and consensus generation.
## Oxford Nanopore Technologies (ONT) data ##
_Mapping:
Medaka (https://nanoporetech.github.io/medaka/ (version 1.2.1)
input: ONT quality processed reads obtained after NanoFilt analysis.
- medaka consensus -m model (default: r941_min_high_g360) –primer (default: empty)
Optional primer removal using iVar follows the same procedure as described before for snippy, being applied before consensus generation.
medaka variant
_VCF filtering:
Mutations are filtered out based on the following user-defined criteria:
Minimum depth of coverage per site (equivalent to –mincov in Illumina pipeline) (default: 30)
Minimum proportion for variant evidence (equivalent to –minfrac in Illumina pipeline) (default: 0.8)
For each mutation, two COVERAGE values are provided in final table output: the depth of unambiguous reads spanning pos +-25 (as provided by medaka variant module) and depth per site as provided by samtools (depth -aa). Values are separated by “/”.
_Consensus generation and mutation annotation (i.e., impact at protein level):
Consensus sequences are generated using bcftools (consensus -s sample.filtered.vcf.gz -f reference.fasta > sample.consensus.fasta) based on the vcf file containing the validated mutations. As for the Illumina pipeline, variant annotation is performed using snpEff 4.1l available with Snippy (see above).
PRIMER CLIPPING: An extra parameter to enable primer removal using iVar (https://genomebiology.biomedcentral.com/articles/10.1186/s13059-018-1618-7) is available within the settings “Mutation detection and consensus generation” section, for both Illumina and ONT. The procedure is an adaptation of the iVar CookBook (https://github.com/andersen-lab/paper_2018_primalseq-ivar/blob/master/cookbook/CookBook.ipynb) and includes these consecutive steps:
Primmer trimming (this adaptation excludes the quality trimming, as this step is done upstream in INSaFLU pipeline)
$ ivar trim -m 0 -q 0 -e -b primers.bed -p samplex.trimmed -i sample.bam
Removal of reads containing minor variants matching primer sequence (only for illumina data)
$ samtools mpileup -A -d 0 -Q 0 sample.trimmed.sorted.bam | ivar consensus -m 0 -n N -p sample.ivar_consensus
$ bwa index -p sample.ivar_consensus sample.ivar_consensus.fa
$ bwa mem -k 5 -T 16 sample.ivar_consensus primer.fasta
$ samtools view -bS -F 4 | samtools sort -o primers_consensus.bam
$ samtools mpileup -A -d 0 –reference sample.ivar_consensus.fa -Q 0 primers_consensus.bam | ivar variants -p primers_consensus -t 0.03
$ bedtools bamtobed -i primers_consensus.bam > primers_consensus.bed
$ ivar getmasked -i primers_consensus.tsv -b primers_consensus.bed -f primer.pair_information.tsv -p primer_mismatchers_indices
$ ivar removereads -i sample.trimmed.sorted.bam -p sample.masked.bam -t primer_mismatchers_indices.txt -b primers.bed
***Users can request trimming of primer sequences of several predefined Primer pool sets, including:
– SARS-CoV-2 Primal Scheme V3 (https://github.com/artic-network/artic-ncov2019/blob/master/primer_schemes/nCoV-2019/V3/nCoV-2019.tsv)
—SARS-CoV-2 Primal Scheme V4.1 (https://github.com/artic-network/artic-ncov2019/tree/master/primer_schemes/nCoV-2019/V4.1)
– Monkeypox Primal Scheme from Welkers, Jonges and van den Ouden (https://www.protocols.io/view/monkeypox-virus-whole-genome-sequencing-using-comb-n2bvj6155lk5/v1)
—Monkeypox Primal Scheme from Chen et al. (https://www.protocols.io/view/monkeypox-virus-multiplexed-pcr-amplicon-sequencin-5qpvob1nbl4o/v2)
Users can upload their own Primer sets (see https://insaflu.readthedocs.io/en/latest/uploading_data.html#uploading-primer-panels)
Masking low coverage regions:
msa_masker.py (https://github.com/rfm-targa/BioinfUtils/blob/master/FASTA/msa_masker.py; kind contribution of Rafael Mamede).
This script substitutes positions with a low depth of coverage in a Multiple Sequence Alignment (MSA) with ‘N’. The depth of coverage value below which the process masks positions is user-selected (see “User-defined parameters”). It will not mask gaps/indels contained in the aligned sequences.
-i: input FASTA file that contains a MAFFT nucleotide alignment enrolling the reference sequence (first sequence of the alignment) and consensus sequence(s) to be masked.
-df: the coverage files (.depth)
-r: define the reference sequence selected by the users (.fasta format)
-c: Positions with a depth value equal or below the value of this argument will be substituted by N (default= “mincov” - 1).
MAPPING VISUALIZATION
Integrative Genomics Viewer (http://software.broadinstitute.org/software/igv/)
inputs: reference file (.fasta); mapping file (.bam; .bai)
Important
INSaFLU allows users to configure key parameters for variant detection and consensus generation. Settings can be user-defined for the whole user account (tab “Settings”), for each project (after project creation) or for individuals samples within a project. When parameters are changed for a given sample within a Project, the sample is automatically re-analysed using the novel parameters and re-inserted in the Project. See details in https://insaflu.readthedocs.io/en/latest/data_analysis.html#user-defined-parameters
Coverage analysis
Description
This module yields a deep analysis of the coverage for each per sample by providing the following data: depth of coverage per nucleotide site, mean depth of coverage per locus, % of locus size covered by at least 1-fold and % of locus size covered by at least a user-defined “mincov” threshold (this parameter is user-selected for a Project or for a given sample within a Project). The latter constitutes the guide for consensus generation, i.e., consensus sequences are exclusively provided for locus fulfilling the criteria of having Y% of their size covered by at least X-fold (X = mincov; Y = minimum horizontal coverage) (see sections “Variant detection and consensus generation” and “User-defined parameters”). Coverage data is provided both in tabular format and interactive plots.
Software version/settings
Note
Script used to generate Coverage statistics:
getCoverage.py (https://github.com/monsanto-pinheiro/getCoverage, by Miguel Pinheiro) (version v1.1; date 15.01.2018)
-i: define the input files, i.e, the coverage files (.depth.gz)
-r: define the reference sequence selected by the users (.fasta format)
-o: defines the output file name (tab-separated value)
Script used to mask low coverage regions
msa_masker.py (https://github.com/rfm-targa/BioinfUtils/blob/master/msa_masker.py; kind contribution of Rafael Mamede)
This script substitutes positions with a low depth of coverage in a Multiple Sequence Alignment (MSA) with ‘N’. The depth of coverage value below which the process masks positions is user-selected (see “User-defined parameters”). It will not mask gaps/indels contained in the aligned sequences.
-i: input FASTA file that contains a MAFFT nucleotide alignment enrolling the reference sequence (first sequence of the alignment) and consensus sequence(s) to be masked.
-df: the coverage files (.depth)
-r: define the reference sequence selected by the users (.fasta format)
-c: Positions with a depth value equal or below the value of this argument will be substituted by N (default= “mincov” - 1).
Alignment/Phylogeny
Description
This module uses filtered nucleotide consensus sequences and performs refined nucleotide/protein sequence alignments and phylogenetic inferences. These outputs are automatically re-build and updated as more samples are added to user-restricted INSaFLU projects, making continuous data integration completely flexible and scalable.
Users can also easily color the phylogenetic tree nodes and/or display colored metadata blocks next to the tree according to any combination of metadata variables, which facilitates the integration of relevant epidemiological and/or clinical data towards an enhanced genome-based pathogen surveillance.
Software version/settings
Note
MAUVE (http://darlinglab.org/mauve/mauve.html) (version 2.4.0; date 15.01.2018)
progressiveMAUVE module (default settings): this algorithm is applied to perform primary draft alignments, and has the particular advantage of automatically concatenating multi-fasta input sequences during whole-genome alignments construction.
input file: filtered nucleotide consensus sequences for each sample, one per each amplicon target (which are , in general, influenza CDSs) and another for the whole-genome sequence (i.e., the set of sequence targeted by the amplicon-based NGS shema, which, in general, is the pool of main 8 influenza CDSs). xmfa to fasta conversion is carried out using “convertAlignment.pl” (https://github.com/lskatz/lyve-SET/blob/master/scripts/convertAlignment.pl
(default settings)
MAFFT (https://mafft.cbrc.jp/alignment/software/) (version 7.313; date 15.01.2018)
For nucleotide alignments:
input file: progressiveMAUVE-derived draft alignments (multifasta format), one per each locus and another for the whole-genome sequence
(default settings)
For amino acid alignments:
–amino: assume the sequences are in amino acid.
FastTree (http://www.microbesonline.org/fasttree/) (version 2.1.10 Double precision; date 15.01.2018)
Double-precision mode: suitable for resolving very-short branch lengths accurately (FastTreeDbl executable)
-nt: defines the input nucleotide alignment, which is a MAFFT-derived refined alignments (multifasta format). Alignments to be run include one per each locus and another for the whole-genome sequence.
—gtr: defines the Generalized time-reversible (GTR) model of nucleotide evolution (CAT approximation with 20 rate categories)
-boot: defines the number resample (-boot 1000)
Seqret EMBOSS tool (http://emboss.sourceforge.net/apps/release/6.6/emboss/apps/) (version 6.6.0.0; date 15.01.2018)
input file: nucleotide alignments in FASTA (.fasta) to be converted in NEXUS (.nex) format
MSAViewer (http://msa.biojs.net/) (latest; date 15.01.2018)
input files: consensus nucleotide alignments for each locus and for the consensus ‘whole-genome’ sequence (upon concatenation of all individual locus); and amino acid alignments for the encoded proteins
Phylocanvas (http://phylocanvas.org/) (version 2.8.1; date 15.01.2018)
input files: phylogenetic tree obtained from each locus-specific nucleotide alignment and from the alignment of the ‘whole-genome’ sequences (upon concatenation of all individual locus)
Metadata visualization tools were built with great contribution from Luís Rita: https://github.com/warcraft12321
Intra-host minor variant detection (and uncovering of putative mixed infections)
Description
This module uses mapping data for the set of samples from each user-restricted INSaFLU project and provides a list of minor intra-host single nucleotide variants (iSNVs), i.e., SNV displaying intra-sample frequency between 1- 50%. This output is automatically re-build and cumulatively updated as more samples are added to each INSaFLU project, making continuous data integration completely flexible and scalable. Plots of the proportion of iSNV at frequency at 1-50% (minor iSNVs) and at frequency 50-90% detected for each sample are also provided as mean to a guide the uncovering of putative mixed infections (exemplified in the Figure). INSaFLU flags samples as “putative mixed infections” based on intra-host SNVs if the following cumulative criteria are fulfilled: the ratio of the number of iSNVs at frequency at 1-50% (minor iSNVs) and 50-90% falls within the range 0,5-2,0 and the sum of the number of these two categories of iSNVs exceeds 20. Alternatively, to account for mixed infections involving extremely different viruses (e.g., A/H3N2 and A/H1N1), the flag is also displayed when the the sum of the two categories of iSNVs exceeds 100, regardless of the first criterion.
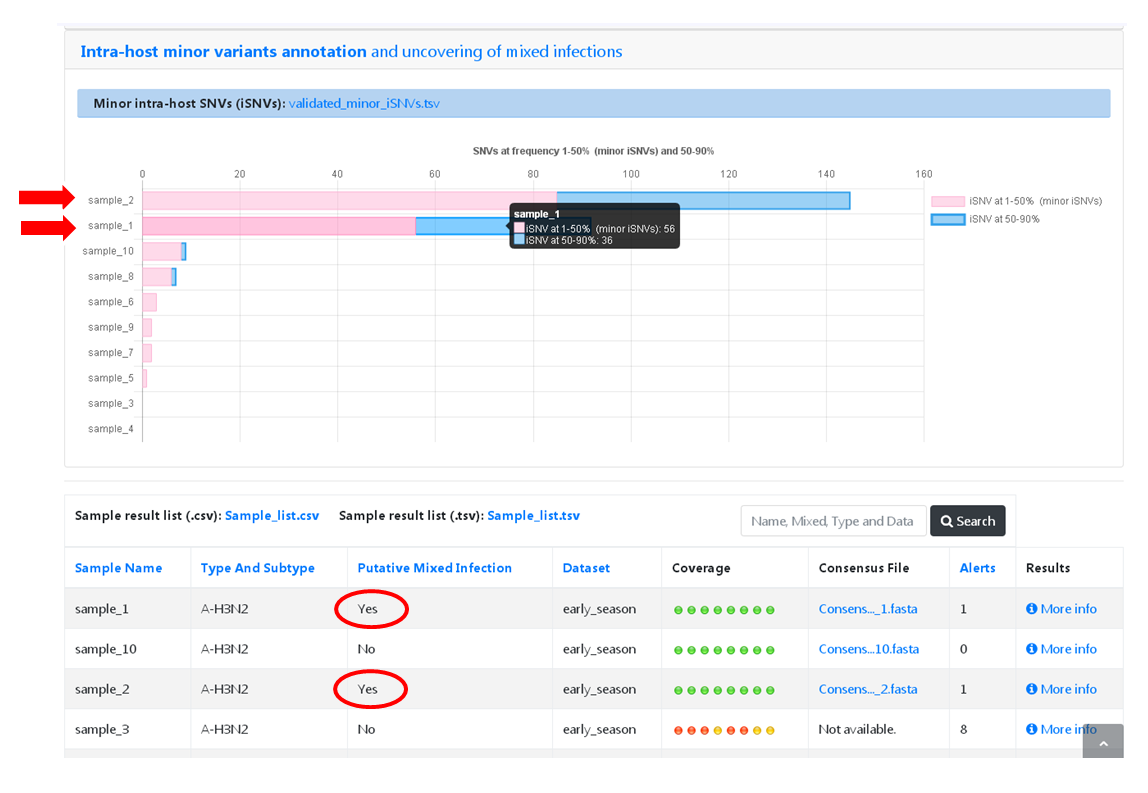
Software version/settings
Note
Freebayes (https://github.com/ekg/freebayes) (version v1.1.0-54-g49413aa; date 15.01.2018)
–min-mapping-quality: excludes read alignments from analysis if they have a mapping quality less than Q (–min-mapping-quality 20)
—min-base-quality: excludes alleles from iSNV analysis if their supporting base quality is less than Q (–min-base-quality 20)
–min-coverage: requires at least 100-fold of coverage to process a site (–min-coverage 100)
—min-alternate-count: require at least 10 reads supporting an alternate allele within a single individual in order to evaluate the position (–min-alternate-count 10)
–min-alternate-fraction: defines the minimum intra-host frequency of the alternate allele to be assumed (–min-alternate-fraction 0.01). This frequency is contingent on the depth of coverage of each processed site since min-alternate-count is set to 10, i.e., the identification of iSNV sites at frequencies of 10%, 2% and 1% is only allowed for sites with depth of coverage of at least 100-fold, 500-fold and 1000-fold, respectively.
Algn2pheno
Description
The align2pheno module in INSaFLU performs the screening of genetic features potentially linked to specific phenotypes. Aln2pheno currently screens SARS-CoV-2 Spike amino acid alignments in each SARS-CoV-2 project against three default “genotype-phenotype” databases: the Carabelli mutations, the COG-UK Antigenic mutations and the Pokay Database (detailed below). Align2pheno reports the repertoire of mutations of interest per sequence and their potential impact on phenotype.
Note
Algn2pheno (https://github.com/insapathogenomics/algn2pheno)
INSaFLU only runs the align2pheno module over Spike amino acid sequences with less than 10% of undefined amino acids (i.e., positions below the coverage cut-off; labelled as “X” in the protein alignments/sequences).
Software and databases versions are provided in a log file in each run.
Databases
Carabelli Database
Description: Database of Spike amino acid mutations in epitope residues listed in Carabelli et al, 2023, 21(3), 162–177, Nat Rev Microbiol (https://doi.org/10.1038/s41579-022-00841-7), Figure 1.
Source: https://github.com/insapathogenomics/algn2pheno/blob/main/tests/DB_SARS_CoV_2_Spike_EpitopeResidues_Carabelli_2023_NatRevMic_Fig1.tsv (prepared and adapted for align2pheno based on https://doi.org/10.1038/s41579-022-00841-7)
Pokay Database
Description: Database of Spike amino acid mutations adapted from the curated database available through the tool Pokay, which includes a comprehensive list of SARS-CoV-2 mutations, and their associated functional impact (e.g., vaccine efficacy, pharmaceutical effectiveness, etc.) collected from literature. Made available by the CSM Center for Health Genomics and Informatics, University of Calgary.
Source: https://github.com/nodrogluap/pokay/tree/master/data
COG-UK Antigenic Mutations Database
Description: Database of Spike amino acid mutations adapted from the COG-UK Antigenic Mutations Database that includes “Spike amino acid replacements reported to confer antigenic change relevant to antibodies, detected in the UK data. The table lists those mutations in the spike gene identified in the UK dataset that have been associated with weaker neutralisation of the virus by convalescent plasma from people who have been infected with SARS-CoV-2, and/or monoclonal antibodies (mAbs) that recognise the SARS-CoV-2 spike protein.” Made available by the COVID-19 Genomics UK (COG-UK) Consortium through the COG-UK/Mutation Explorer.
Nextstrain Datasets
Description
This module allows the creation of datasets for further in-depth phylogenetic analysis using Nextstrain (https://docs.nextstrain.org/en/latest/index.html). This provides an advanced vizualization and exploration of phylogenetic and genomic data, allowing the integration of geographic and temporal data and further user-provided metadata.
Currently, INSaFLU allows the creation of Datasets using virus-specific Nextstrain builds (seasonal Influenza, SARS-CoV-2 and Monkeypox) as well as a “generic” build that can be used for any pathogen.
More details here: https://github.com/INSaFLU/nextstrain_builds
Builds
Seasonal influenza
INSaFLU allows running four Nexstrain builds for the seasonal influenza (A/H3N2, A/H1N1/, B/Victoria and B/Yamagata), which are simplified versions of the Influenza Nextstrain builds available at https://github.com/nextstrain/seasonal-flu
So far, influenza analyses are restricted to the Hemagglutinn (HA) coding gene. The reference HA sequences used for site (nucleotide / amino acid) numbering in the output JSON files are:
H1N1PDM: A/California/07/2009(H1N1) (https://www.ncbi.nlm.nih.gov/nuccore/CY121680.1/)
H3N2: A/Beijing/32/1992 (https://www.ncbi.nlm.nih.gov/nuccore/U26830.1/)
VIC: Influenza B virus (B/Hong Kong/02/1993) (https://www.ncbi.nlm.nih.gov/nuccore/CY018813.1/)
YAM: Influenza B virus (B/Singapore/11/1994) (https://www.ncbi.nlm.nih.gov/nuccore/CY019707.1/)
This build uses the following software versions: nextstrain-cli (3.2.4), augur (15.0.2), nextalign (1.11.0), nextclade (1.11.0)
Avian influenza
INSaFLU allows running Nexstrain builds for the avian influenza (A/H5N1), which are a simplified version of the Nextstrain builds available at https://github.com/nextstrain/avian-flu
Nextstrain avian influenza can be launched for each of the 8 segments. The reference sequences used for site (nucleotide / amino acid) numbering in the output JSON files are:
HA: Influenza A virus (A/Goose/Guangdong/1/96(H5N1)) hemagglutinin (HA) (https://www.ncbi.nlm.nih.gov/nuccore/AF144305.1/)
NA: Influenza A virus (A/Goose/Guangdong/1/96(H5N1)) neuraminidase (NA) (https://www.ncbi.nlm.nih.gov/nuccore/AF144304.1)
PB2: Influenza A virus (A/Goose/Guangdong/1/96(H5N1)) polymerase (PB2) (https://www.ncbi.nlm.nih.gov/nuccore/AF144300.1)
PB1: Influenza A virus (A/Goose/Guangdong/1/96(H5N1)) polymerase (PB1) (https://www.ncbi.nlm.nih.gov/nuccore/AF144301.1)
PA: Influenza A virus (A/Goose/Guangdong/1/96(H5N1)) polymerase (PA) (https://www.ncbi.nlm.nih.gov/nuccore/AF144302.1)
MP: Influenza A virus (A/Goose/Guangdong/1/96(H5N1)) matrix proteins M1 and M2 (M) gene (https://www.ncbi.nlm.nih.gov/nuccore/AF144306.1)
NS: Influenza A virus (A/Goose/Guangdong/1/96(H5N1)) nonstructural proteins NS1 and NS2 (NS) gene (https://www.ncbi.nlm.nih.gov/nuccore/AF144307.1)
This build uses the following software versions: nextstrain-cli (3.2.4), augur (15.0.2), nextalign (1.11.0), nextclade (1.11.0)
SARS-CoV-2
This build is a simplified version of the SARS-CoV-2 Nextstrain build available at https://github.com/nextstrain/ncov
The reference genome used for site (nucleotide / amino acid) numbering and genome structure in the output JSON files is:
Wuhan-Hu-1/2019 (https://www.ncbi.nlm.nih.gov/nuccore/MN908947)
This build uses the following software versions: nextstrain-cli (3.2.4), augur (15.0.2), nextalign (1.11.0), nextclade (1.11.0)
Monkeypox virus
This build is a simplified version of the Monkeypox virus Nextstrain build available at https://github.com/nextstrain/monkeypox
The reference genome used for site (nucleotide / amino acid) numbering and genome structure in the output JSON files is:
MPXV-M5312_HM12_Rivers (https://www.ncbi.nlm.nih.gov/nuccore/NC_063383)
This build uses the following software versions: biopython (1.74), nextstrain-cli (4.2.0), augur (17.1.0), auspice (2.38.0), nextalign (2.5.0), nextclade (2.5.0), epiweeks (2.1.4), pangolin (2.4.2), pangolearn (2021.05.27), seqkit (2.3.0), tsv-utils (v2.2.0).
Respiratory Syncytial Virus (RSV)
This build is a simplified version of the RSV virus Nextstrain build available at https://github.com/nextstrain/rsv
The reference genome used for site (nucleotide / amino acid) numbering and genome structure in the output JSON files is:
RSV A: A/England/397/2017 (GISAID ID EPI_ISL_412866)
RSV B: B/Australia/VIC-RCH056/2019 (GISAID ID EPI_ISL_1653999)
This build uses the following software versions: augur (20.0), auspice (2.42), nextalign (2.9.1), nextclade (2.9.1), epiweeks (2.1.4).
Generic
This build is a simplified version of the Nextstrain build available at https://github.com/nextstrain/zika
This generic build uses as reference sequence (as tree root and for mutation annotation) one of the reference sequences of the projects included in the Nextstrain dataset.
Currently, the generic build does not generate a Time-Resolved Tree. To do this you need to select the Generic with TimeTree option.
This build uses the following software versions: nextstrain-cli (3.2.4), augur (15.0.2), nextalign (1.11.0), nextclade (1.11.0)
Generic with TimeTree
This build is similar to the Generic build, but it also builds a time tree, inferring a mutation rate from the sample dates. Like in the Generic build, one reference is required to align the dataset consensus sequences. Nonetheless, unlike in the Generic build, the reference is not specifically defined as the root, but the root is inferred from the data instead. To make use of this build, you need to accurately specify dates associated with each sample.
Important
To take advantage of temporal and geographical features of Nextstrain, please make sure you provide:
“collection date” for all samples added to Nextstrain datasets. If no collection date is provided, INSaFLU will automatically insert the date of the analysis as the “collection date”, which might (considerably) bias (or even break) the time-scale trees generated for influenza, SARS-CoV-2 and Monkeypox.
“latitude” and “longitude” AND/OR “region”, “country”, “division” and/or “location” columns in the metadata. These values will be screened against a vast database of “latitude and longitude” coordinates (https://github.com/INSaFLU/nextstrain_builds/blob/main/generic/config/lat_longs.tsv) to geographically place the sequences in the Nextstrain map.
To add/update the Nextstrain metadata of a given Dataset, click in “Metadata for Nextstrain”, download the previous table, update it with new data and upload it. Then, click in the “hourglass” icon to Rebuild the Nexstrain outputs. Note: you can also add/update the metadata of sequences previously obtained with INSaFLU (i.e., consensus sequences coming from the “Projects” module), please follow these instructions: https://insaflu.readthedocs.io/en/latest/uploading_data.html#updating-sample-metadata (this option is not available for external sequences).
Metagenomics virus detection
The TELEVIR bioinformatics component of INSaFLU is a modular pipeline for the identification of viral sequences in metagenomic data (both Illumina and ONT data).
It is composed of these main steps (detailed below):
Read quality analysis and improvement [optional]
Extra-filtering [optional].
Viral Enrichment [optional].
Host Depletion [optional].
De novo assembly of the reads [optional].
- Identification of the viral sequences.
Using reads.
Using contigs (if assembled).
Using several reference databases.
Selection of viral TAXID and representative genome sequences for confirmatory re-mapping
Remapping of the viral sequences against selected reference genome sequences.
Reporting
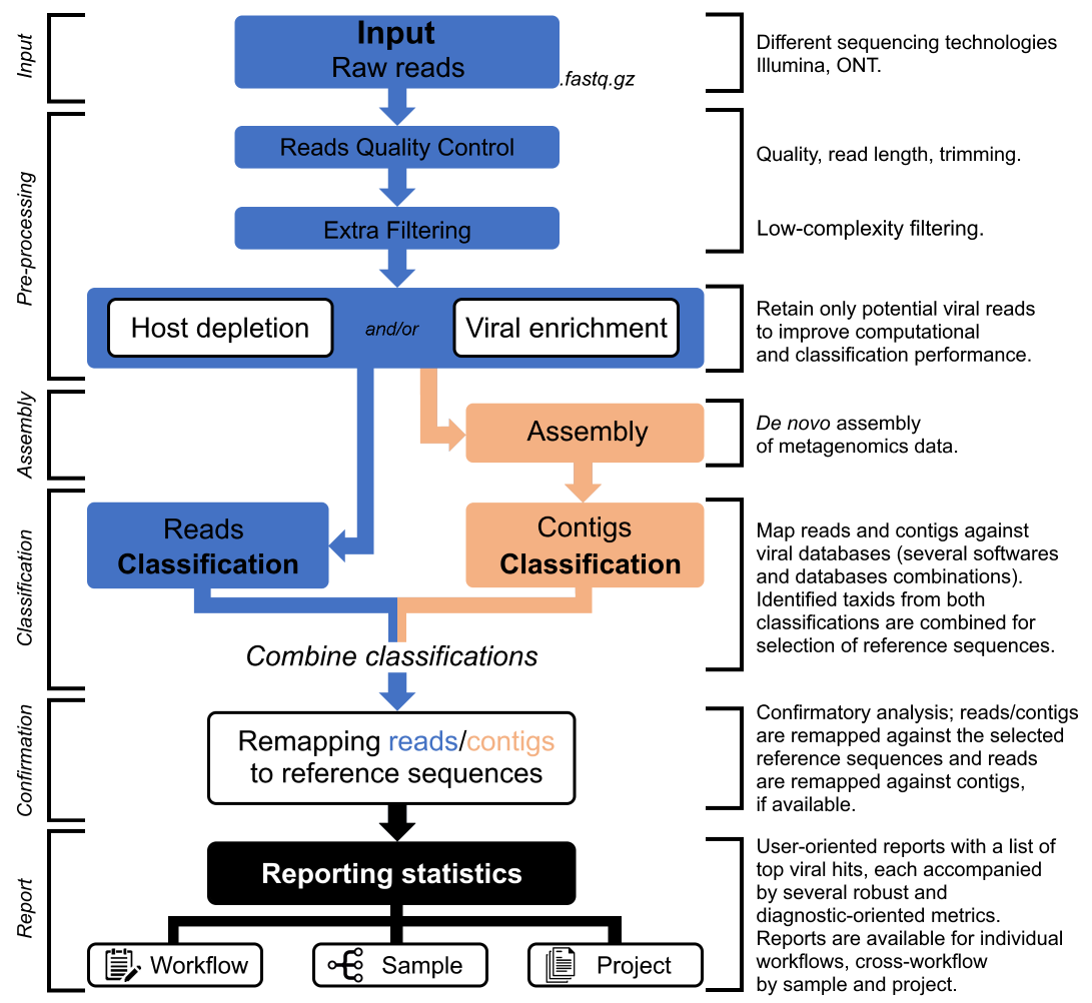
Simplified illustration of the main steps of the modular INSaFLU-TELEVIR bioinformatics pipeline for metagenomics virus diagnostics.
Find below the description of the main bioinformatics steps (Description, Current software versions and settings)
Read quality analysis and improvement
Description
This step takes the input single- or paired-end reads (fastq.gz format; ONT or Illumina) and produces quality processed reads, as well as quality control reports for each file, before and after this step. This module overlaps the two components (virus detection and surveillance) of the platform.
Software version/settings
Note
## ILLUMINA data ##
FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) (version 0.11.5; date 15.01.2018)
input: single- or paired-end reads (fastq.gz format) (e.g., sample_L001_R1_001.fastq.gz and sample_L001_R2_001.fastq.gz for Illumina technology reads)
—nogroup option: all reports will show data for every base in the read.
Trimmomatic (http://www.usadellab.org/cms/index.php?page=trimmomatic) (version 0.27; date 15.01.2018)
input: single- or paired-end reads (fastq.gz format) (e.g., sample_L001_R1_001.fastq.gz and sample_L001_R2_001.fastq.gz for Illumina paired-end reads)
ILLUMINACLIP: To clip adapters from the input file using user-specified adapter sequences. ILLUMINACLIP:<ADAPTER_FILE>:3:30:10:6:true
HEADCROP: Cut the specified number of bases from the start of the read. Range: [0:100]. If value equal to 0 this parameter is excluded.
CROP: Cut the read to a specified length. Range: [0:400]. If value equal to 0 this parameter is excluded.
SLIDINGWINDOW: perform a sliding window trimming, cutting once the average quality within the window falls below a threshold (default: SLIDINGWINDOW:5:20, where 5 refers to window and 20 to the minimum average quality)
LEADING: cut bases off the start of a read, if below a threshold quality (default: LEADING:3). This will allow discarding bases with very quality or N bases (quality score of 2 or less).
TRAILING: cut bases off the end of a read, if below a threshold quality (default: TRAILING:3). This will allow discarding bases with very quality or N bases (quality score of 2 or less).
MINLEN: drop the read if it is below a specified length (MINLEN:35)
TOPHRED33: Convert quality scores to Phred-33
## Oxford Nanopore Technologies (ONT) data ##
NanoStat (https://github.com/wdecoster/nanostat) (version 1.4.0)
input: ONT reads (fastq.gz format)
NanoFilt (https://github.com/wdecoster/nanofilt) (version 2.6.0)
-q (QUALITY): Filter on a minimum average read quality score. Range: [5:30] (default: 10)
-l (LENGTH): Filter on a minimum read length. Range: [50:1000]. (default: 50)
–headcrop: Trim n nucleotides from start of read. Range: [1:1000]. If value equal to 0 this parameter is excluded. (default: 70)
–tailcrop: Trim n nucleotides from end of read. Range: [1:1000]. If value equal to 0 this parameter is excluded. (default: 70)
–maxlength: Set a maximum read length. Range: [100:50000]. If value equal to 0 this parameter is excluded. (default: 0)
RabbitQC (https://github.com/ZekunYin/RabbitQC) (version 0.0.1)**
input: ONT reads (fastq.gz format) pre- and post- quality improvement with NanoFilt
Files between 50 - 300 MB are downsized to ~50 MB before analysis by randomly sampling reads using fastq-sample from fastq-tools package https://github.com/dcjones/fastq-tools (developed by Daniel C. Jones dcjones@cs.washington.edu)
Important
INSaFLU allows users to configure key parameters for reads quality analysis in the tab “Settings”.
Please check your settings before uploading new samples to your account.
See details in https://insaflu.readthedocs.io/en/latest/data_analysis.html#user-defined-parameters
Extra Filtering
Description
This step remove reads enriched in low complexity regions (e.g., homopolymeric tracts or repeat regions), which are a common source of false-positive bioinformatics hits**. This step is directly performed using over raw reads (if QC was turned OFF) or quality processed reads (if QC was turned ON).
Software
Host depletion
Description
This step removes potential host reads based on reference-based mapping against host genome sequence(s). Mapped reads are treated as potential host reads and removed. If the Extra filetring and Viral enrichment steps were turned OFF, host depletion will be directly performed over raw reads (if QC was turned OFF) or quality processed reads (if QC was turned ON).
Software
BWA (https://github.com/lh3/bwa)
Minimap2 (https://github.com/lh3/minimap2)
Host reference sequences*
Human reference genome hg38 - NCBI accid GCA_000001405.15
De novo Assembly
Description
This step performs de novo assembly using reads retained after the “Viral enrichment” and/or “Host depletion” steps. If the latter steps were turned OFF, assembly will be directly performed using raw reads (if QC was turned OFF) or quality processed reads (if QC was turned ON).
Software
SPAdes (https://github.com/ablab/spades)
Reporting
TELEVIR reports are generated per Workflow, per Sample (combining non-redundant hits detected across workflows) and per Project (combining several samples), with a decreasing level of detail.
The workflows culminate in user-oriented reports with a list of top viral hits, each accompanied by several robust and diagnostic-oriented metrics, statistics and visualizations, provided as (interactive) tables (intermediate and final reports), graphs (e.g., coverage plots, Integrative Genomics Viewer visualization, Assembly to reference dotplot) and multiple downloadable output files (e.g., list of the software parameters, reads/contigs classification reports, mapped reads/contigs identified per each virus; reference sequences, etc).
See the description of the reports and outputs here: https://insaflu.readthedocs.io/en/latest/metagenomics_virus_detection.html#televir-output-visualization-and-download
Sorting
In order to simplify the final reports (per Sample and per Workflow) and facilitate the identification of potential false positive hits (often arising from cross-mapping with true positive hits), sample-specific viral references are grouped together by mapping affinity, as measured by shared mapped reads.
In summary, reference hits selected for remapping are first pooled across sample workflows, excluding references with no mapped reads. A neighbor-joining tree based on shared reads is then constructed. A maximum of 100000 reads are used after filtering out reads shared by 5% of references or less. A filter is then applied on the inner nodes of the tree that considers the distribution of shared reads as summarized by two statistics:
private_reads : the proportion of reads found to map only to descendents of that node.
pairwise_min : the minimum reciprocal proportion of reads mapped between every pair of descendents (i.e. all samples must share at least X% (user-defined) of their reads with another sample among descendents of the same node).
Thresholds for these two statistics are defined beforehand: private_reads is set by the user through the parameter “–r-overlap” (defaults to 50 %); pairwise_min is a TELEVIR constant set to 5 %. After filtering, nodes are sorted by the total number of reads mapped to their descendents. Finally, references (tree leaves) are mapped to the filtered inner nodes and sorted accordingly. Orphaned leaves and references with no mapped reads are appended last.
Warnings and Flags
TELEVIR reports provide specific Warnings for bioinformatics “artifacts” commonly yielding false-positive taxid assignments. Calculations depend on the flag-type, a user defined variable (TELEVIR Settings – Reporting – Final Report - Flagging and Sorting – –flag-type, default: viruses), and target broad characteristics of main input types.
Two flag-types currently exist for viruses (oriented to shotgun viral metagenomics) and probes (oriented for probe-based NGS target panels)
## Flag-type “viruses” (default)
“Likely False Positive”: when most reads map to a very small region of the reference sequence, i.e., hits with high “DepthC” but low “Depth” and low “Cov (%)”. Flagged for hits with DepthC / Depth > 10 and Cov (%) < 5%.
“Vestigial Mapping”: when only a vestigial amount of reads (<= 2) mapped.
## Flag-type “probes”
“Likely False Positive”: When the reference genome is not sufficiently covered as a function of the number of the proportion of Windows Covered, calculated as above. Flagged for hits with Windows Covered <= 50 % (calculated from the fraction presented)
“Vestigial Mapping”: when only a vestigial amount of reads (<= 2) mapped.
User-defined parameters (UNDER CONSTRUCTION)
INSaFLU allows turning ON/OFF specific modules and user-defined configuration of key parameters for reads quality analysis, INSaFLU and TELEVIR projects. Settings can be user-defined for the whole user account (tab “Settings”), for each project (just after project creation) or for individual samples within a project (click in the “Magic wand” icon).
Please choose your settings before uploading new samples to your account.
Example:

Read quality analysis and improvement control (QC)
##ILLUMINA / Ion Torrent data##
Users can change the following Trimmomatic settings (see http://www.usadellab.org/cms/index.php?page=trimmomatic):
ILLUMINACLIP: To clip the Illumina adapters from the input file using the adapter sequences. ILLUMINACLIP:<ADAPTER_FILE>:3:30:10:6:true (default: Not apply)
HEADCROP: <length> Cut the specified number of bases from the start of the read. Range: [0:100]. If value equal to 0 this parameter is excluded. (default = 0)
CROP:<length> Cut the read to a specified length. Range: [0:400]. If value equal to 0 this parameter is excluded. (default = 0)
SLIDINGWINDOW:<windowSize> specifies the number of bases to average across Range: [3:50]. (default = 5)
SLIDINGWINDOW:<requiredQuality> specifies the average quality required Range: [10:100]. (default = 20)
LEADING:<quality> Remove low quality bases from the beginning. Range: [0:100]. If value equal to 0 this parameter is excluded. (default = 3)
TRAILING:<quality> Remove low quality bases from the end. Range: [0:100]. If value equal to 0 this parameter is excluded. (default = 3)
MINLEN:<length> This module removes reads that fall below the specified minimal length. Range: [5:500]. (default = 35)
NOTE: “Trimming occurs in the order which the parameters are listed”
## Oxford Nanopore Technologies (ONT) data ##
Users can change the following NanoFilt settings (see: https://github.com/wdecoster/nanofilt)
QUALITY: Filter on a minimum average read quality score. Range: [5:30] (default: 10)
LENGTH: Filter on a minimum read length. Range: [50:1000]. (default: 50)
HEADCROP: Trim n nucleotides from start of read. Range: [1:1000]. If value equal to 0 this parameter is excluded. (default: 70)
TAILCROP: Trim n nucleotides from end of read. Range: [1:1000]. If value equal to 0 this parameter is excluded. (default: 70)
MAXLENGTH: Set a maximum read length. Range: [100:50000]. If value equal to 0 this parameter is excluded. (default: 0)
Mapping, Variant Calling
##ILLUMINA / Ion Torrent data##
Users can change the following Snippy settings (see also https://github.com/tseemann/snippy):
–mapqual: minimum mapping quality to accept in variant calling (default = 20)
–mincov: minimum number of reads covering a site to be considered (default = 10)
–minfrac: minimum proportion of reads which must differ from the reference, so that the variant is assumed in the consensus sequence (default = 0.51)
## Oxford Nanopore Technologies (ONT) data ##
Users can change the following settings:
Medaka model (default: r941_min_high_g360) (see: https://nanoporetech.github.io/medaka/)
Minimum depth of coverage per site (equivalent to –mincov in Illumina pipeline) (default: 30)
Minimum proportion for variant evidence (equivalent to –minfrac in Illumina pipeline) (default: 0.8). Note: medaka-derived mutations with frequencies below the user-defined “minfrac” will be masked with an “N”.
Consensus generation (horizontal coverage cut-off) and Masking
Users can select the Minimum percentage of horizontal coverage to generate consensus. This threshold indicates the Minimum percentage of locus horizontal coverage with depth of coverage equal or above –mincov (see Mapping settings) to generate a consensus sequence for a given locus. Range: [50:100] (default = 70)
In Projects setting, users can also mask (i.e., put Ns) specific regions (or sites) of the consensus sequences for all (or individual) samples within a given Project. This feature is especially useful for masking the start/end of the sequences or known error-prone nucleotide sites.
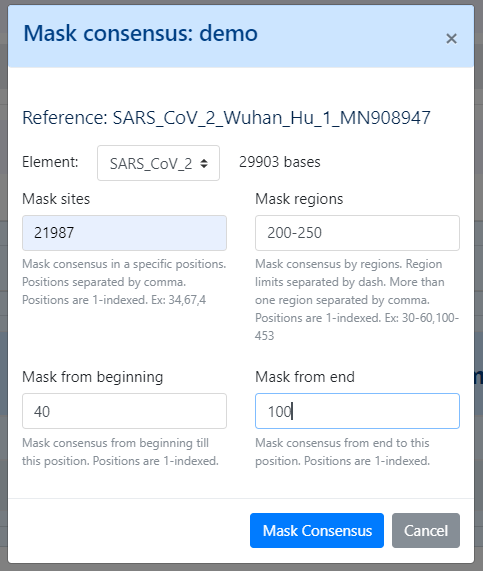
Masking summary:
Undefined nucleotides (NNN) are automatically placed in: i) low coverage regions (i.e., regions with coverage below –mincov); ii) regions (or sites) selected to be masked by the user (in Projects settings); iii) for ONT data, medaka-derived mutations with frequencies below the user-defined “minfrac” (i.e. Minimum proportion for variant evidence).