Uploading or Updating data
INSaFLU needs:
NGS data (fastq reads) (mandatory)
Sample metadata (to link each sample to the respective NGS data) (mandatory)
Reference data (additional user-restricted reference sequences) (optional)
Uploading Sample metadata and NGS data
Samples’s metadata and respective NGS data (single-end or paired-end reads in fastq.gz format obtained through widely used technologies, such as Illumina or Ion Torrent) can be uploaded to INSaFLU as a batch (option 1) or individually (option 2):
# Option 1 (Batch)
A. Go to Samples menu and choose Add Multiple Samples / Load new file.
Examples of template table files are provided in this menu.
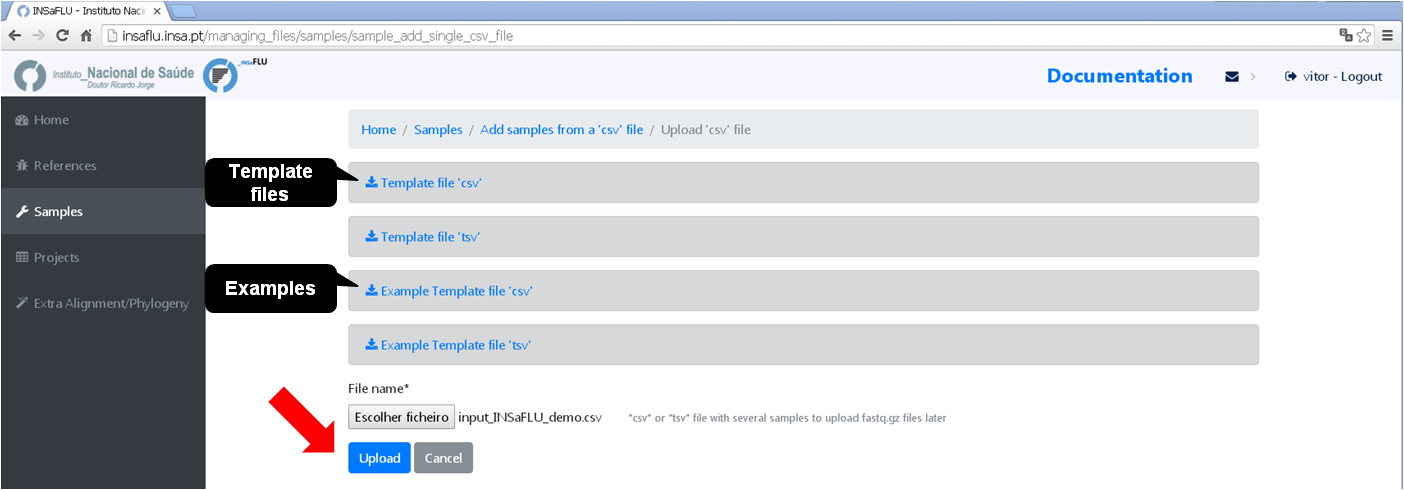
See here how to upload the metadata table:
Important
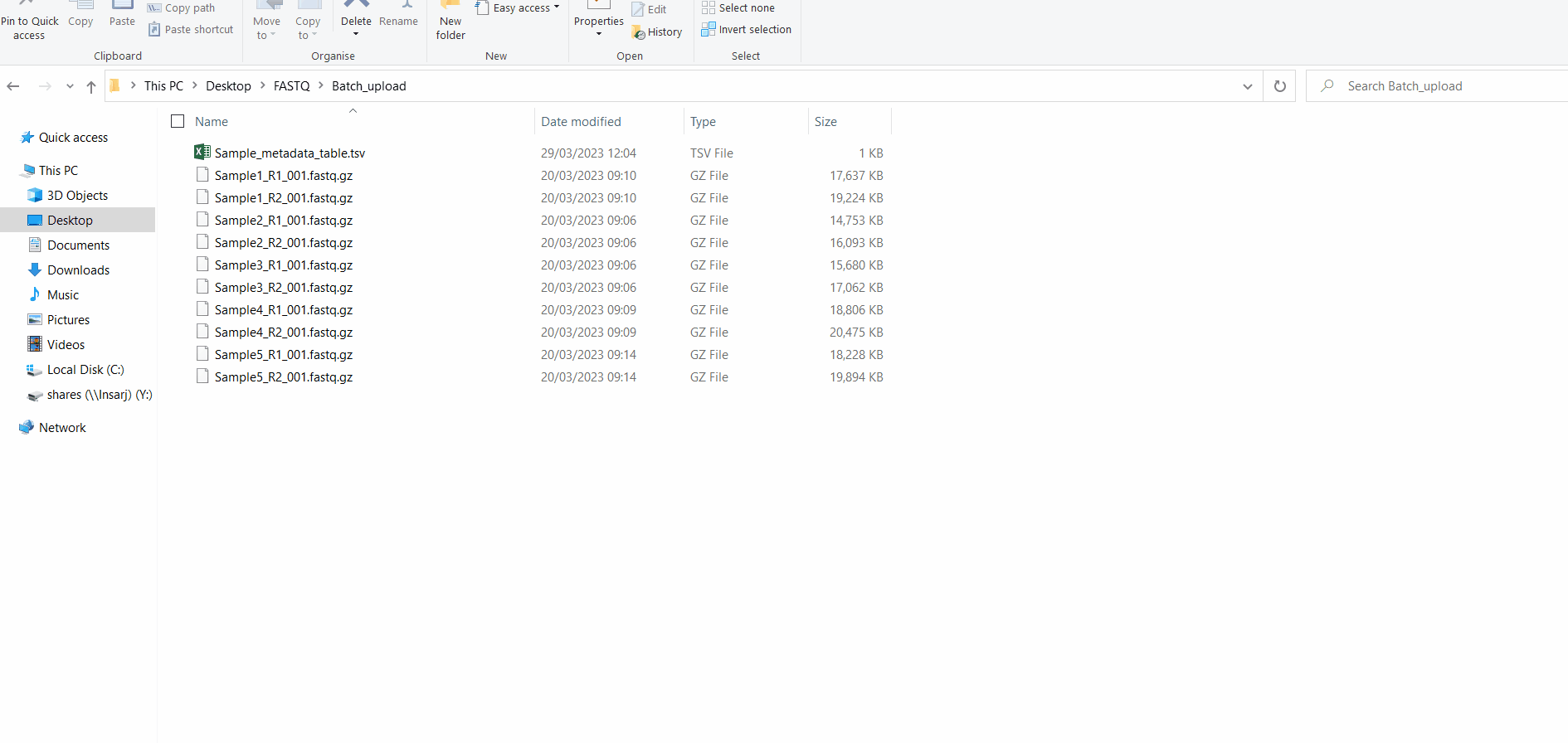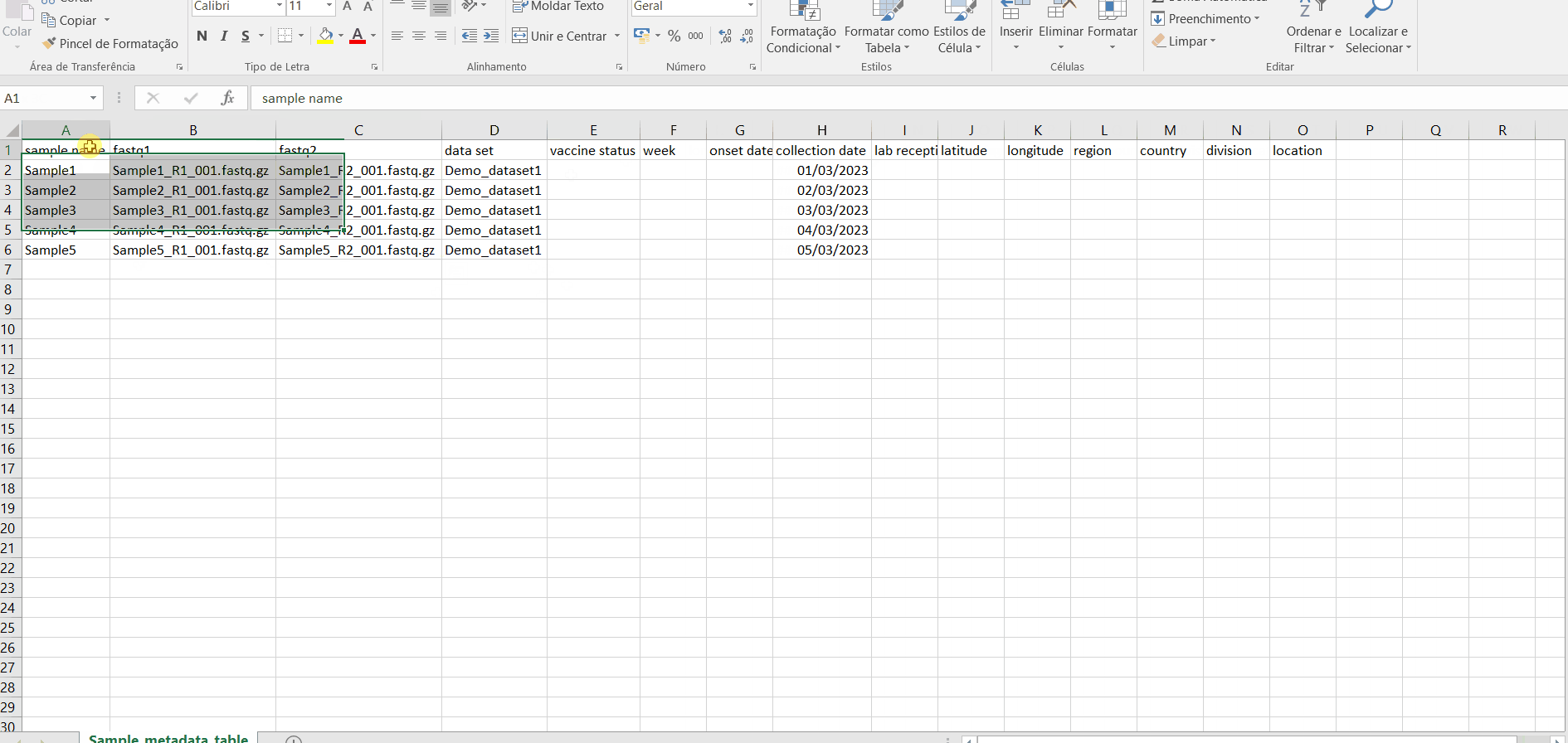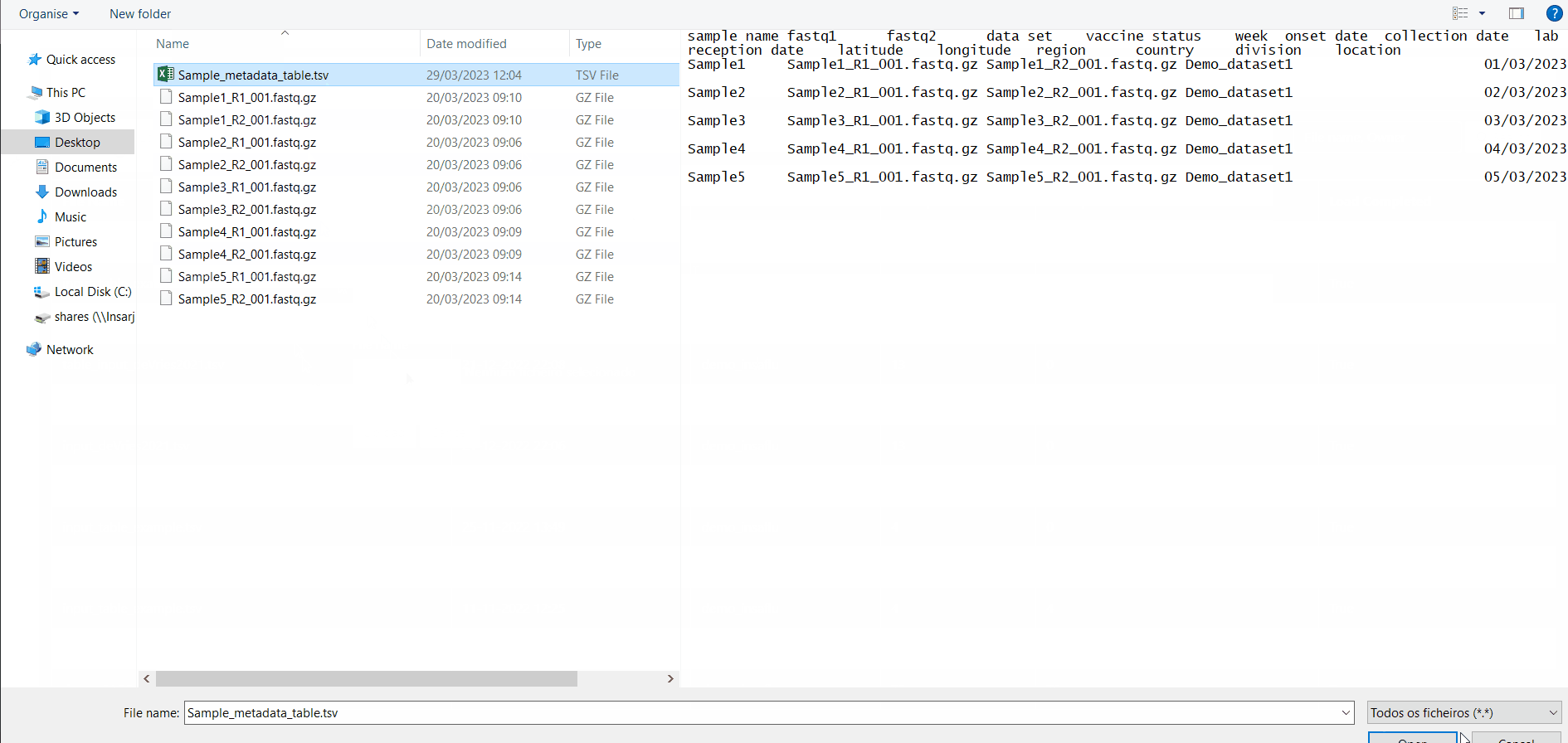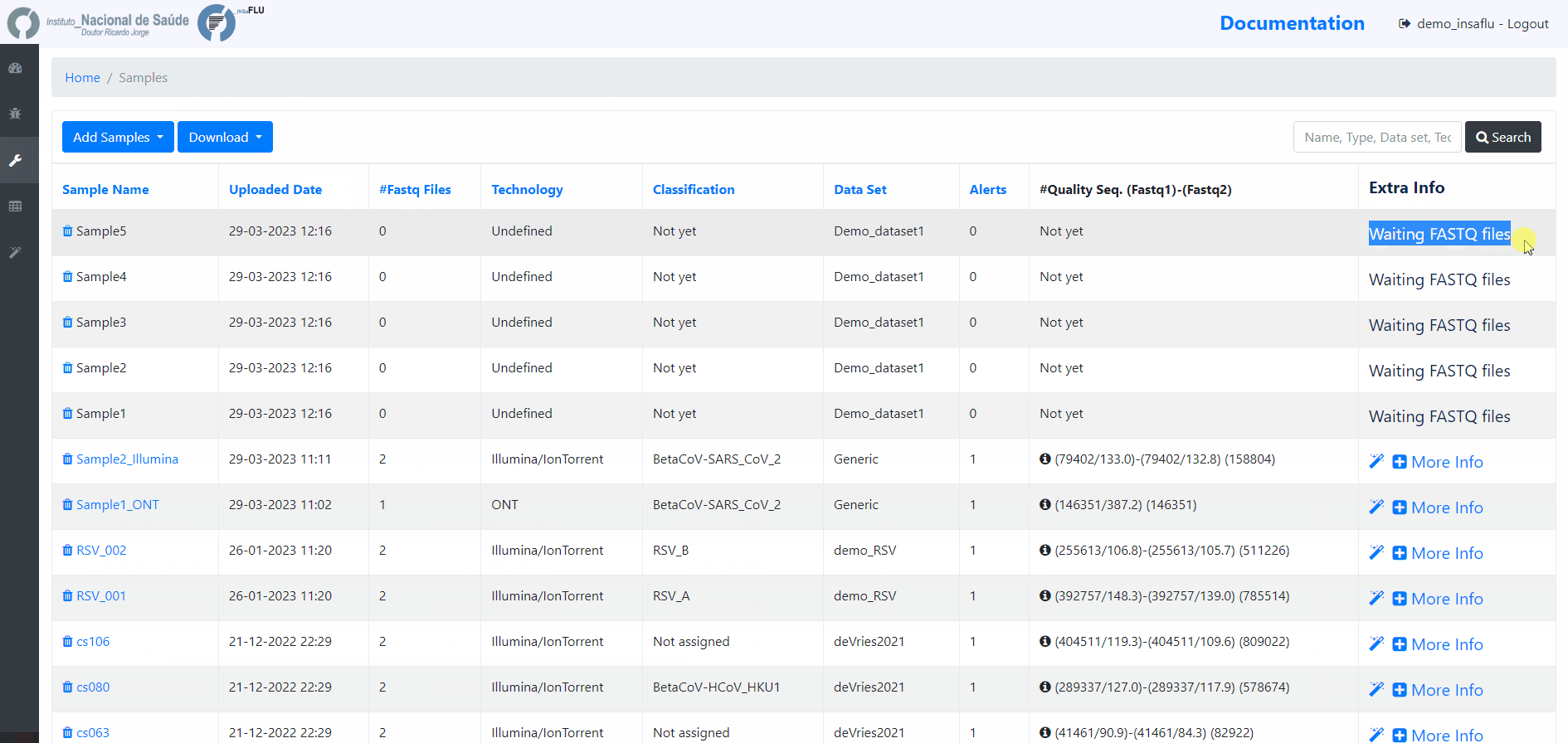
Sample metadata should be a comma-separated value (.csv) or tab-separated value (.tsv or .txt) table containing the columns “sample name”, “fastq1” and “fastq2” (mandatory columns to fulfill; NOTE: fastq2 is exceptionally not fulfilled only for single-end data) as well these additional variables (that may not be fulfilled): “data set”, ”vaccine status”, ”week”, ”onset date”, ”collection date”, ”lab reception date”, ”latitude”, ”longitude”. If you include data for latitude/longitude and/or country, region, division and/or location, these will be used to geographically locate your samples in the Nextstrain Datasets
Users are encouraged to include any other columns with metadata variables to be associated with samples (see advantages below).
The table will be locked until you submit the NGS reads corresponding to the new samples. In case you decide not to proceed with the upload, the table can be unlocked by clicking in “Unlock last file”.
Important
Samples names in your account must be unique and only numbers, letters and underscores are allowed.
Fastq.gz file names must be complete (including extension) and match the ones from the files that you are going to upload later.
Advantages of uploading enriched Sample metadata tables
The template file contains the following additional variables, which commonly constitute the minimal metadata collected during seasonal influenza surveillance: “data set”, ”vaccine status”, ”week”, ”onset date”, ”collection date”, ”lab reception date”, ”latitude” and ”longitude”. (The “data set” column will help you to organize samples by specific groups, e.g., “early_season”, “peak”, etc). Users are also recommended to include data for for latitude/longitude and/or country, region, division and/or location, as these fields will allow geographically locate your samples in the Nextstrain Datasets. Still, users are encouraged to include any other columns with metadata variables to be associated with samples.
Uploading tables enriched with multiple metadata variables will allow you to easily color the phylogenetic tree nodes (and/or add colored metadata blocks to the tree) according to any combination of metadata variables, which largely facilitates the visualization and exploration of your data.
Also, it has the clear advantage of allowing the subsequent upload of the samples list of each project (output: “Sample_list.csv” or “Sample_list.tsv”) along to the standardized and multi-format outputs of INSaFLU projects (alignments/trees) to other platforms for phylogenetic data visualization and/or phylogeographical analysis, such as:
auspice (https://auspice.us/), which takes JSON files or sample metadata (in tsv-separated format) plus a phylogenetic tree (“.nwk” format).
Phandango (https://jameshadfield.github.io/phandango/#/), which runs sample metadata (csv-separated format) plus a phylogenetic tree (“.tree” format);
Microreact (https://microreact.org/), which takes sample metadata (in csv-separated format) plus a phylogenetic tree (“.nwk” format).
Note
To take full advantage of particular platforms, such Microreact, which provides a framework where associated data (geographical, temporal, phenotypic or epidemiological) can be easily explored together with phylogenetic data, users may need to specify additional columns in the table (e.g. specifying “year”, “month” and “day” columns, along with “latitude” and “longitude” allows Microreact to automatically integrate temporal and geolocation data; ). Also, for colour/shape definitions, you will need to create extra columns (see Microreact instructions here: https://microreact.org/instructions).
It is worthnoting that, for Microreact, depending on the tree (whole-genome or locus-specific trees) that you use (which can include a distinct set of viruses), you will additionally need to match the number of identifiers within the id column to the number of viruses in each tree.
B. Go to Samples menu and choose Add Fastq Files
Go to your fastq.gz directory and simultaneously upload multiple fastq.gz files, which are automatically linked to the corresponding samples.
Reads (fastq.gz) can be selected from the multi-select dialog box or just DRAG & DROPPED.

Important
Please confirm in the “Home / Samples / Add Multiple Samples” page whether all uploaded samples ((#Samples Processed) have been processed (#Samples Processed), i.e., if all samples in the metadata table were attached to the respective reads. if not, go to the “Home / Samples / Add Fastq” page, delete the files not attached and upload them again.
Fastq.gz files that are not attached to any sample can be deleted by clicking in “Remove all files not processed”.
Note
How to merge several ONT fastq/fastq.gz files into a single ONT fastq/fastq.gz on “Windows” or “MAC” (see video below):
Download one of these files from https://github.com/INSaFLU/INSaFLU/raw/master/files_helpful/ (check the README)
concat_fastq (for fastq files)
concat_fastqgz (for fastq.gz)
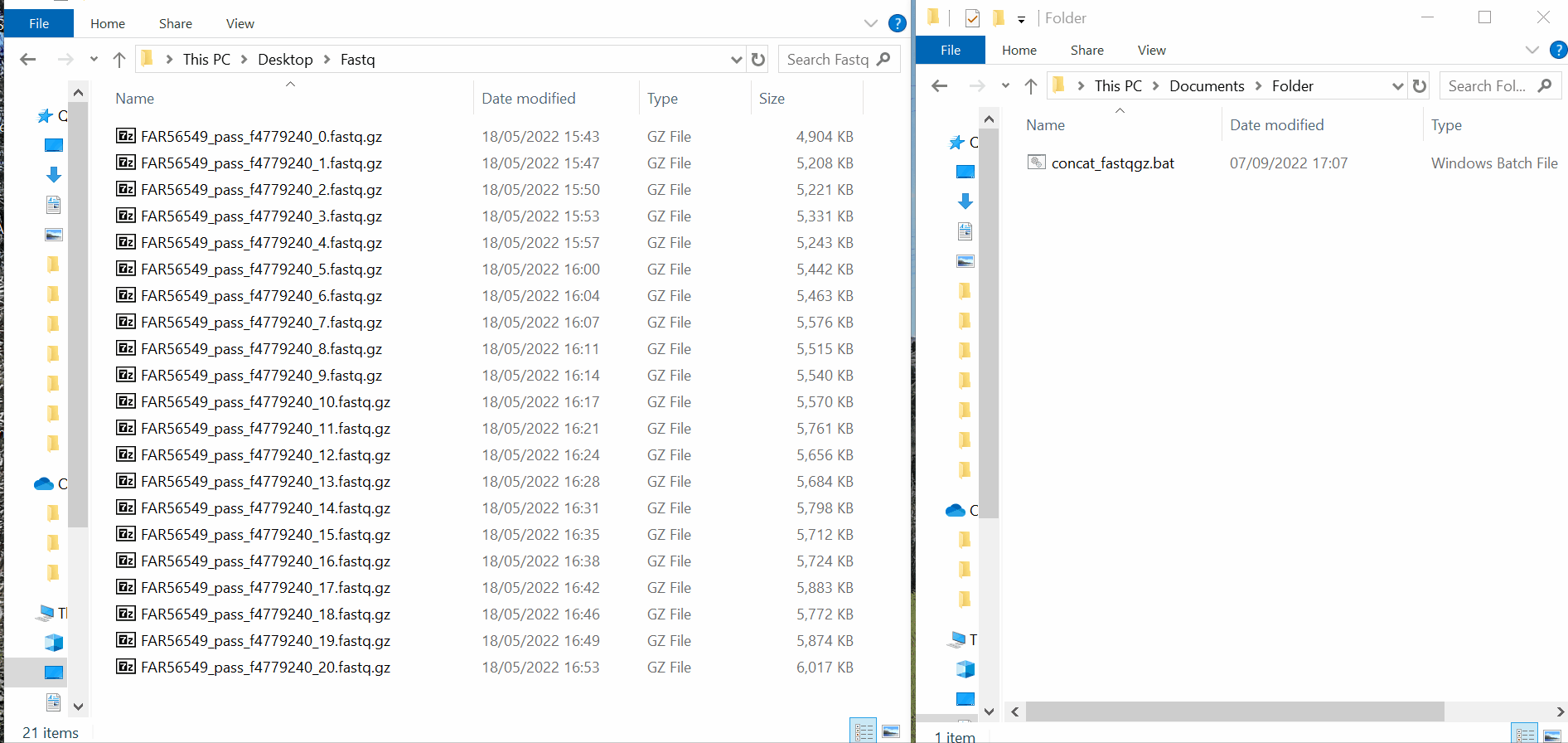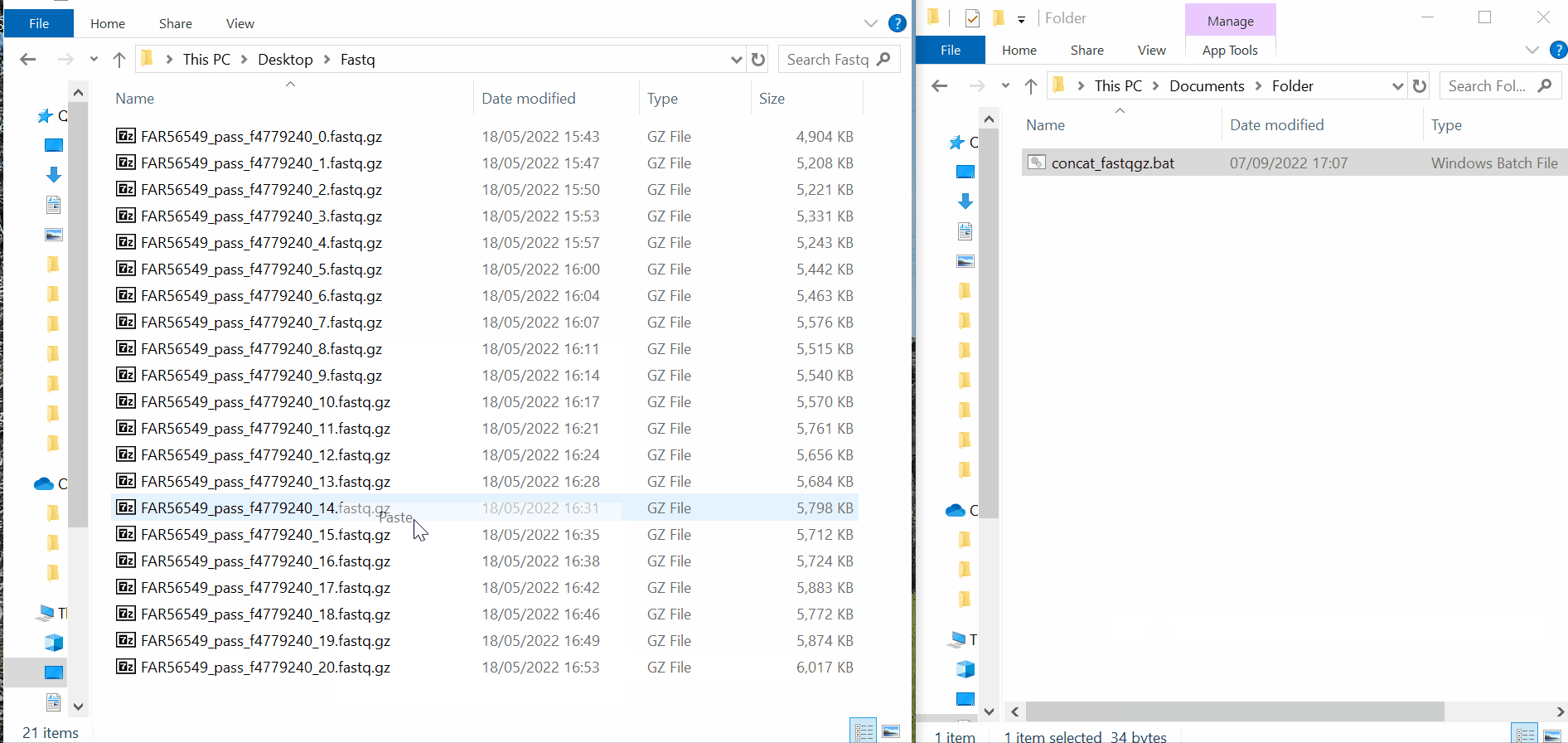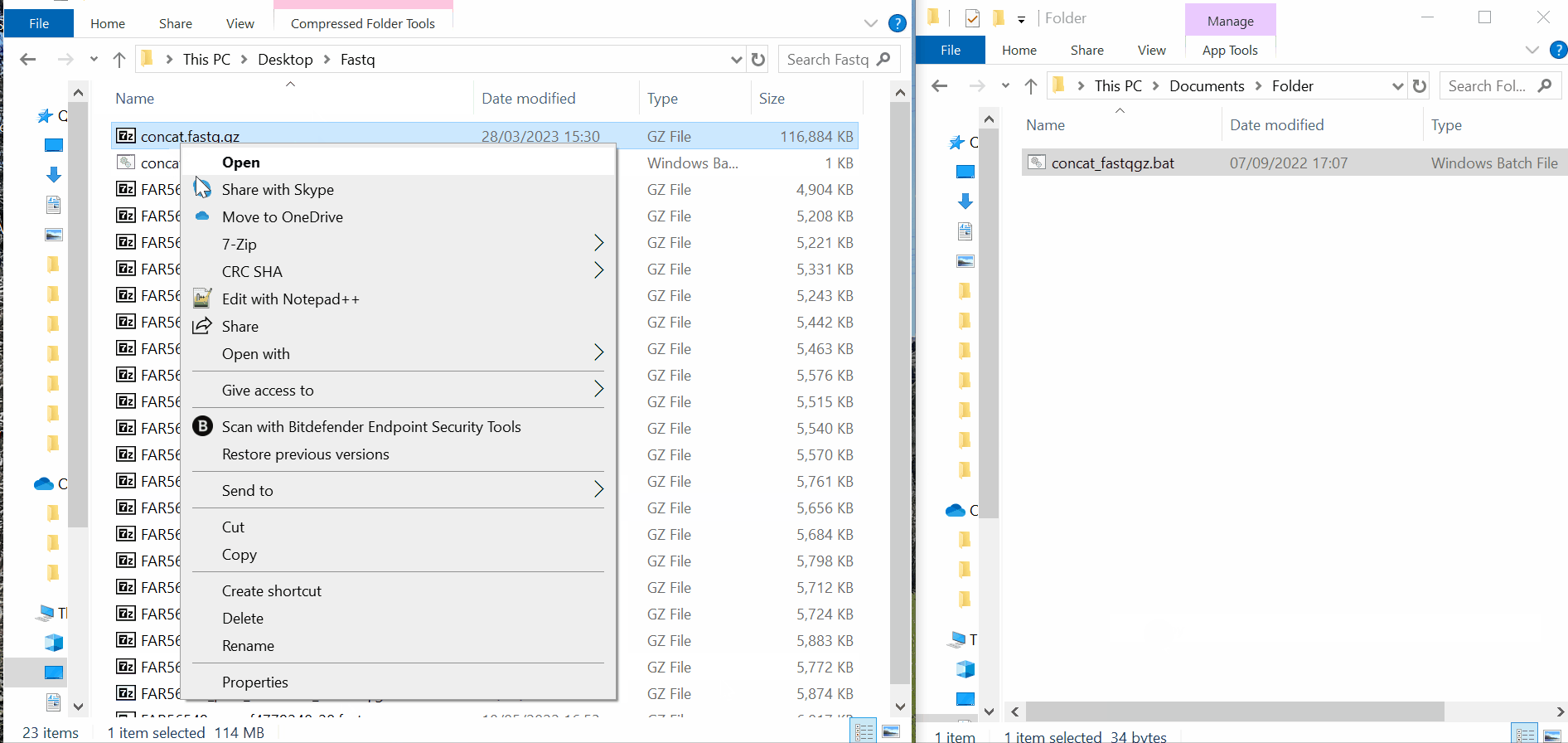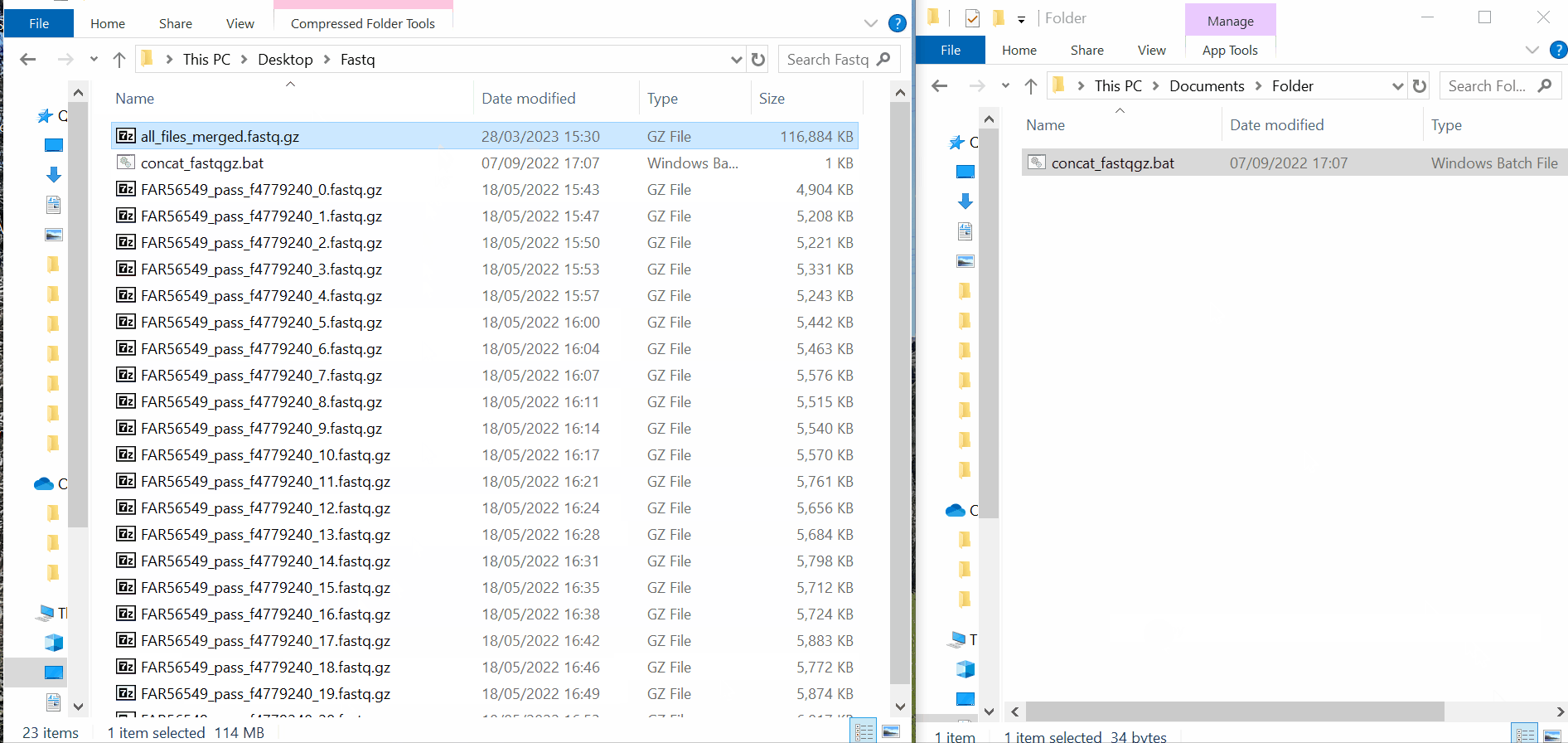
Create a folder where you only put the fastq/fastq.gz you wish to concatenate
Copy&paste the adequate “.bat” (Windows) or “.command” (MAC) file (whether you have fastq or fastq.gz) to the same folder and double-click on the file
This will automatically create a single file named “concat.fastq.gz” (or “concat.fastq”) inside the same folder. You can then rename this file as needed. (Note that this will not eliminate or change the original fastq inside the folder.)
Important
Original reads (i.e., the fastq.gz files uploaded) will be deleted (normally after 10 days) of their upload. In fact, after quality analysis and improvement, the INSaFLU pipeline does not use those original reads for any other downstream analysis (all quality reports and derived quality processed reads are kept available for download).
# Option 2 (Individual)
A. Go to Samples menu and choose Add One Sample
Here you can upload each sample at the time (including associated metadata and NGS data).
Example for a ONT sample:

Example for a Illumina (paired-end) sample:

Important
Original reads (i.e., the fastq.gz files uploaded) will be deleted (normally after 10 days) of their upload. In fact, after quality analysis and improvement, the INSaFLU pipeline does not use those original reads for any other downstream analysis (all quality reports and derived quality processed reads are kept available for download).
Note
How to merge several ONT fastq/fastq.gz files into a single ONT fastq/fastq.gz on “Windows” or “MAC” (see video below):
Download one of these files from https://github.com/INSaFLU/INSaFLU/raw/master/files_helpful/ (check the README)
concat_fastq (for fastq files)
concat_fastqgz (for fastq.gz)
Create a folder where you only put the fastq/fastq.gz you wish to concatenate
Copy&paste the adequate “.bat” (Windows) or “.command” (MAC) file (whether you have fastq or fastq.gz) to the same folder and double-click on the file
This will automatically create a single file named “concat.fastq.gz” (or “concat.fastq”) inside the same folder. You can then rename this file as needed. (Note that this will not eliminate or change the original fastq inside the folder.)
Important
This “individual” upload option does not allow the user to add additional metadata fields (contrarily to batch upload; see next section to lear how to update the sample metadata).
Original reads (i.e., the fastq.gz files uploaded) will be deleted (normally after 10 days) of their upload. In fact, after quality analysis and improvement, the INSaFLU pipeline does not use those original reads for any other downstream analysis (all quality reports and derived quality processed reads are kept available for download).
Updating Sample metadata
You can add/update the Samples’s metadata at any time by simply uploading a table comma-separated (.csv) or tab-separated (.tsv or .txt) table with the updated data (a template file is provided), as follows:
Go to Samples menu and choose Update metadata, then Load a new file, using one of the provided template files.
Important
The table for Sample metadata update should contain the columns “sample name” (exactly corresponding to the samples to be updated), as well these additional variables (that may not be fulfilled): “data set”, ”vaccine status”, ”week”, ”onset date”, ”collection date”, ”lab reception date”, ”latitude”, ”longitude”, “country, region”, “division” and/or “location”. If you include data for “latitude” and “longitude” AND/OR country, region, division and/or location, these will be used to geographically locate your samples in the Nextstrain Datasets. Still, users are encouraged to include any other columns with metadata variables to be associated with samples (see advantages above).
Note
You cannot update the fastq files. To replace the fastq files associated with a given sample, you need to delete the respective sample and upload all data (metadata plus fastq) again, i.e., create a new sample. You must ensure that the sample to be deleted is not inserted in any Project before deleting it.
Samples names in your account must be unique and only numbers, letters and underscores are allowed.
You can add/update the Nextstrain metadata of a given Dataset by clicking in “Metadata for Nextstrain”, download the previous table, update it with new data and upload it again. Then, you need to click in the “hourglass” icon to Rebuild the Nextstrain outputs. When the new JSON file is generated, the updated metadata can then be drag-and drop to auspice.us after the JSON file to update the displayed metadata.
Notes: - This update option is exclusive of each Dataset. - To add/update the metadata associated with consensus sequences derived from read data in INSaFLU Projects module, you can also add/update the metadata following these instructions of the previous point “A” (this option is not available for “external” consensus sequences).
Uploading Reference data
INSAFLU References
INSaFLU needs reference sequence files to be used for reference-based mapping.
In References menu, INSaFLU provides a set of ready-to-use reference sequences, all publicly available at NCBI (or made available under permission of authors), currently including:
post-pandemic (2009) vaccine/reference influenza A(H1N1)pdm2009, A(H3N2) and B viruses (from both Northern and Southern hemispheres);
representative virus of multiple combinations of HA/NA subtypes (i.e., H1N1, H2N2, H5N1, H7N9, etc)
SARS-CoV-2 reference genome sequence (Wuhan-Hu-1; NCBI accession MN908947)
Mpox reference sequences
RSV references sequences
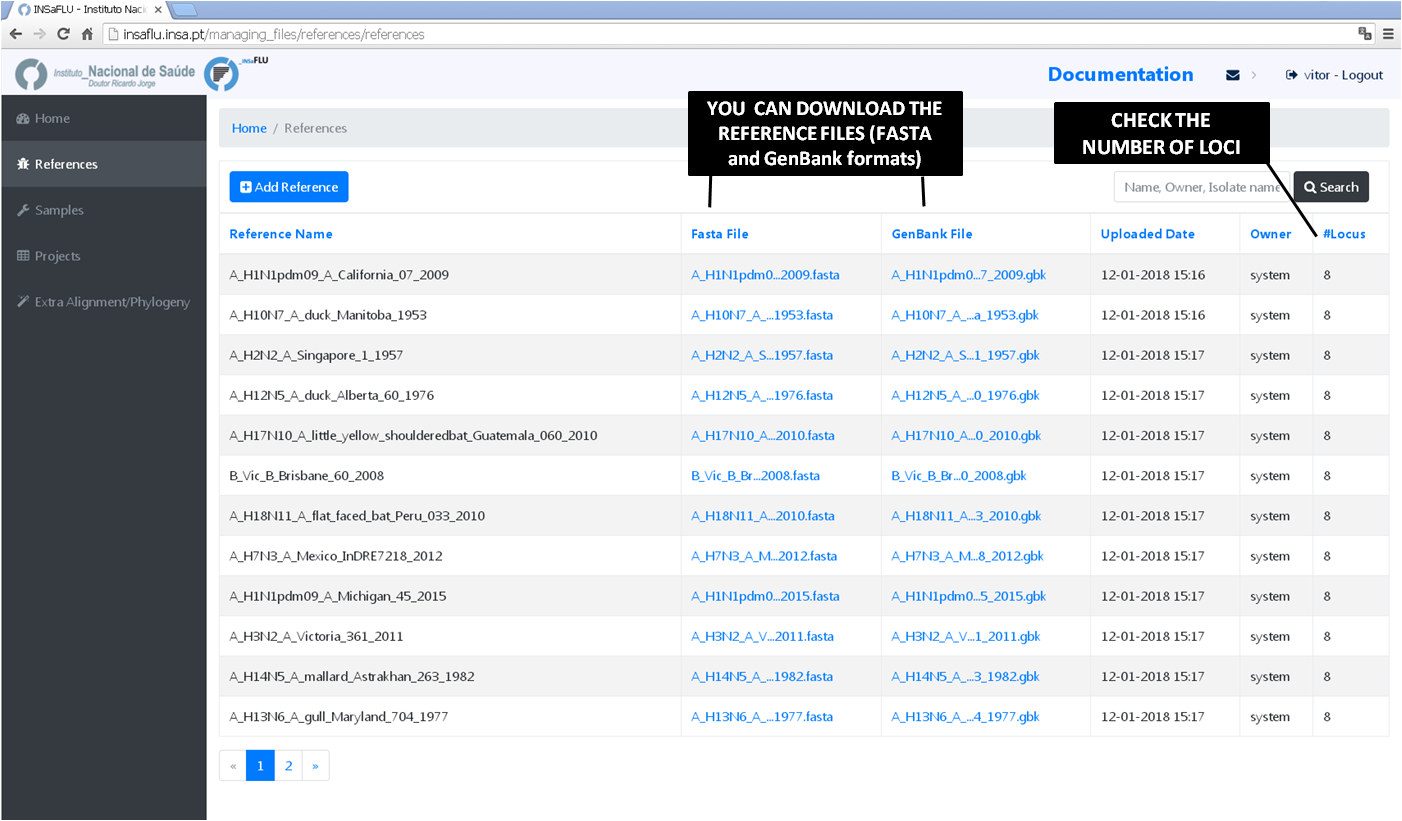
The current list of reference sequences, including loci size and NCBI accession numbers is provided here:
INSaFLU_current_REFERENCE_DATABASE_07_03_2023.xlsx
NOTE: The default seasonal influenza reference files (FASTA and GenBank formats) have been prepared to fit amplicon-based schemas capturing the whole CDS of the main eight genes of influenza virus (PB2, PB1, PA, HA, NP, NA, M and NS), such as the wet-lab pre-NGS protocol for influenza whole genome amplification adapted from a RT-PCR assay described by Zhou and colleagues (Zhou et al, 2009, for Influenza A; and Zhou et al, 2014, for Influenza B; Zhou and Wentworth, 2012).
You can download the suggested protocol here: Suggested_RT_PCR_assay_for_influenza_WGS.pdf
Important
NO FURTHER ACTIONS ARE NEEDED if you are using the suggested wet-lab pre-NGS protocol and you want to compare your sequences against a reference available at INSaFLU database.
However, you may need to UPLOAD additional reference files to the user-restricted reference database. For instance, you may need to upload the A/H3N2 vaccine reference sequence for the season 2017/2018 (A/Hong Kong/4801/2014 virus), which is not freely available.
To upload additional reference sequences, GO TO References MENU and CHOOSE Add Reference
You can eitheir:
Upload a FASTA + GENBANK file

Note
Your genbank file must be compatible with INSaFLU. ## See below a GUIDE to generate additional reference sequences
Upload a FASTA file
In this case, INSaFLU will automatically generate an annotation in GenBabk format*

Note
Important notes - upload new References:
Multi-FASTA files to be uploaded will typically contain the set of reference sequences that constitute the influenza “whole-genome” sequence of a particular virus (e.g, the combination of the traditional 8 amplicons targeting the 8 eight influenza RNA segments). Still, you are free to upload references files including a specific panel of segments/genes (e.g, segments 4 and 6, which encode the surface proteins HA and NA, respectively)
Each individual sequence of the multi-FASTA file should ideally have the size of each “intra-amplicon” target sequence that you capture by each one of the RT-PCR amplicons. Otherwise, you will get regions with no or low coverage (these will be masked with undefined bases NNN according to the user-defined coverage thresholds).
INSaFLU automatically provide a draft annotation of the uploaded multi-FASTA sequences upon submission, but you can also upload (preferentially) a respective multi-GenBank file, e.g., downloaded from GenBank or generated with tools like https://www.ncbi.nlm.nih.gov/genomes/FLU/annotation/. In both case, please be aware that the GenBank file should be modified if you intend to have the annotation compatible with the “HA1 numbering” (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4229193/) commonly applied for seasonal influenza (see instructions below).
## See below a GUIDE to generate additional reference sequences
TELEVIR References
The TELEVIR module relies on reference databases, or their indices, for metagenomics analysis. Validation of metagenomics hits in turn consists in analysing and comparing the alignment of reads and contigs to the reference sequences.
In this context, the INSaFLU TELEVIR provides the utilities to allow the following: - Explore the TELEVIR reference data base - e.g. to confirm that references of interest are available. - Upload new references to the TELEVIR reference database - Note: these references will only be availble for Investigatory Mapping (see TELEVIR documentation for more details). - Create Reference Panels - to help allow streamlined analysis of specific viruses or virus families.
TELEVIR Reference utilities are accessed via the TELEVIR References menu.
Upload new reference sequences
The TELEVIR Reference Files page displays the current reference databases available for use in the TELEVIR module, both provided and user-uploaded.
To upload new reference sequences, click on the Upload Reference Panel button. This will require two files:
A FASTA.GZ file containing reference sequences, multiple sequences allowed - limited in size Max total sequence length: 5242800 bp. Max FASTA file size: 100.0 MB.
A metadata .tsv file containing accession ID, Taxon ID and Description columns, with specific column names. A template is provided.
References Search
The TELEVIR Reference Files page also provides a search function to allow users to search the reference database for specific sequences.
By default this page displays user submitted references. It is possible to create an INSaFLU reference from TELEVIR references. Genbank files will be automatically generated.
Panels
The panels page displays the current reference panels available for use in the TELEVIR module, both provided and user-uploaded.
To create a new reference panel, click on the Create Reference Panel button. References to include in a panel can be selected from the reference databases. Alternatively, the user can select a user-uploaded reference panel to add references in bulk.
GUIDE TO GENERATE ADDITIONAL REFERENCE SEQUENCES
Please take this guide into account when generating additional reference sequences.
(Multi-)FASTA files to be upload typically contain the reference sequence(s) that constitute the “whole-genome” sequence of a particular virus (e.g, the combination of the traditional 8 amplicons targeting the 8 eight influenza RNA segments in a MULTI-FASTA file or the reference genome sequence for SARS-CoV2 in a Single FASTA file). Each individual sequence should ideally have the precise size of each “intra-amplicon” target sequence that you capture by each one of the RT-PCR amplicons. Otherwise, you will obtain regions with no or low coverage (these will be masked with undefined bases NNN according to the user-defined coverage thresholds)
You may generate your (multi-)FASTA files in order to fit your amplicon schema by simply adjusting the whole-genome sequences available for download at INSaFLU or at influenza-specific sequence repositories, such as the Influenza Research Database (https://www.fludb.org), NCBI Influenza Virus Resource (https://www.ncbi.nlm.nih.gov/genomes/FLU/Database/nph-select.cgi?go=database) and EpiFLU/GISAID (https://www.gisaid.org/).
An easy way to handle/generate (multi-)FASTA files is by opening a text file (e.g., NOTEPAD) and paste individual sequences after each header line. The FASTA IDs (after the ‘>’ character) represent the individual sequence names. For the sake of simplicity, you may designate each sequence as 1, 2, 3, 4, 5, 6 , 7 and 8 (see example), following the traditional influenza segments order (keeping this numerical order is advisable). At the end, you just have to save the (multi-)FASTA file as “.fasta”. Please avoid symbols or blank spaces in the file names.
example:
A_H3N2_A_Perth_16_2009.fastaINSaFLU automatically provide a draft annotation of the uploaded multi-FASTA sequences upon submission, but you can also upload (preferentially) a respective multi-GenBank file, e.g., downloaded from GenBank or generated with tools like https://www.ncbi.nlm.nih.gov/genomes/FLU/annotation/. If you upload FASTA and respective GenBank files that have been downloaded from NCBI, please make sure that FASTA ID(s) (after the ‘>’ character) match the name/number that appears in the LOCUS and ACCESSION lines of the GenBank file.. Be also aware that the GenBank file should be modified if you intend to have the annotation compatible with the “HA1 numbering” (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4229193/) commonly applied for seasonal influenza (see instructions below).
INSaFLU requires reference sequences exclusively composed by non-degenerate bases (i.e. A, T, C, or G). As such, please ensure that all degenerated bases (e.g., R, Y, M, K, S and W) are replaced by non-degenerate sequences before uploading. The choice of the base used in the replacement (e.g., “A” or “G” when replacing an “R”) has no impact on the analysis. It simply means that mutations falling in the replaced nucleotide position will be reported taking into account the reference base selected.
IMPORTANT:
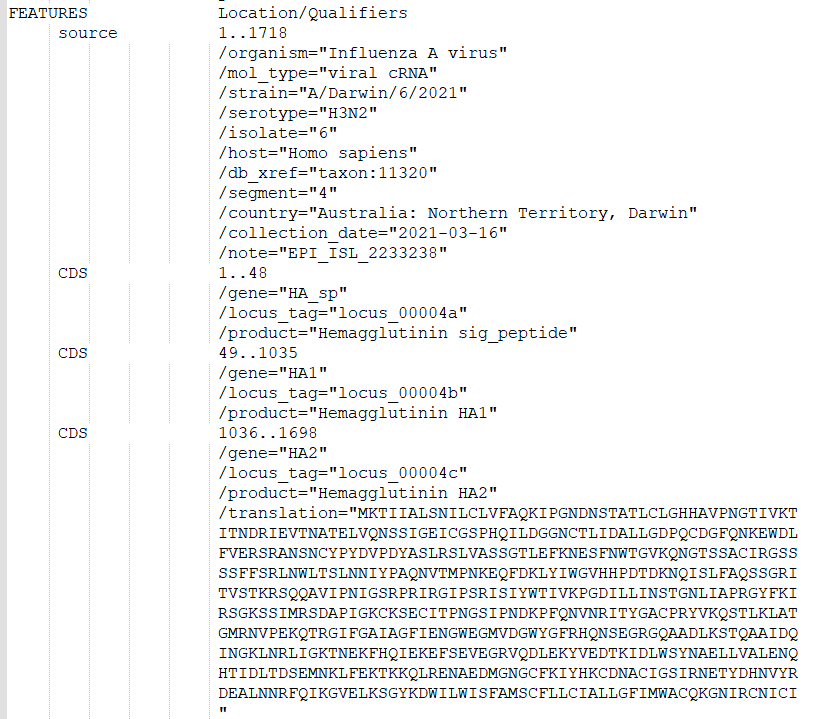
The influenza GenBank files available at NCBI are not compatible with the “HA1 numbering” (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4229193/) commonly applied for seasonal influenza. As such, in order to get mutations annotated for each peptide (signal peptide, HA1 and HA2 peptides), instead of the full-protein, make sure you modified the GenBank file as follows:
THESE ORIGINAL LINES in THE GENBANK FILE ….

…SHOULD BE REPLACED BY:
Uploading Primer Panels
INSaFLU enables the use of Primer Panels for read filtering in the preprocessing phase (see https://insaflu.readthedocs.io/en/latest/bioinformatics_pipeline.html#read-quality-analysis-and-improvement), or for filtering in the variant detection and consensus generation phase (see the Primer Clipping section in https://insaflu.readthedocs.io/en/latest/bioinformatics_pipeline.html#variant-detection-and-consensus-generation).
There are some pre-loaded primer panels available in INSaFLU, but users can upload their own.

To upload a primer panel, click on the Add Primer, and provide a fasta and primer pair files, as indicated:

You can see examples of files in the INSaFLU db: https://github.com/INSaFLU/INSaFLU/tree/master/static/db/primers
Explore your Sample and Reference databases
Samples menu displays all information for all loaded samples (Samples’ names in your account must be unique).
Upon submission, INSaFLU automatically updates samples’ information with reads quality and typing data (automate bioinformatics pipeline modules “Read quality analysis and improvement” and Type and sub-type detection”; see Data analysis in the Documentation).
Upon deployment of TELEVIR workflows on any sample, any additionally processed data (i.e. Entropy filterered, Enriched, Host depleted, see Metagenomics virus detection section) will be automatically added to the sample database.
Just explore the “More info” icon next to each sample.

References menu displays all information for all reference sequences available at your confidential session.
Both FASTA and GenBank files can be downloaded by clicking on the displayed links.
References:
Zhou B, Donnelly ME, Scholes DT, St George K, Hatta M, Kawaoka Y, Wentworth DE. 2009. Single-reaction genomic amplification accelerates sequencing and vaccine production for classical and Swine origin human influenza a viruses. J Virol, 83:10309-13.
Zhou B, Lin X, Wang W, Halpin RA, Bera J, Stockwell TB, Barr IG, Wentworth DE. 2014. Universal influenza B virus genomic amplification facilitates sequencing, diagnostics, and reverse genetics. J Clin Microbiol, 52:1330-1337.
Zhou B, Wentworth DE. 2012. Influenza A virus molecular virology techniques. Methods Mol Biol, 865:175-92.